14.8.4. steps.API_1.solver
Implementation of serial simulation solvers.
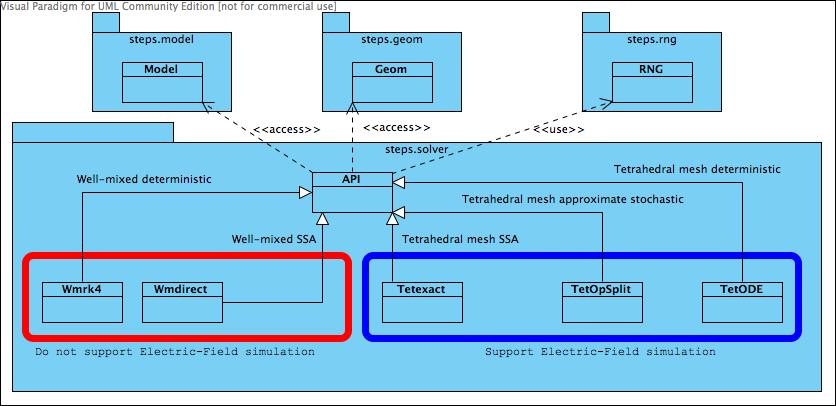
Class diagram for steps.API_1.solver namespace.
Each solver is a partial or full implementation of the STEPS solver API. At the moment STEPS implements four different serial solvers (and one parallel solver in steps.API_1.mpi.solver namespace).
steps.API_1.solver.Wmrk4 implements a well-mixed, deterministic solver
based on the Runge–Kutta method.
steps.API_1.solver.Wmdirect implements a stochastic, well-mixed solver
based on Gillespie’s Direct SSA Method.
steps.API_1.solver.Wmrssa implements a stochastic, well-mixed solver
based on the Rejection SSA method (Thanh V, Zunino R, Priami C (n.d.) On the rejection-based algorithm for simulation and analysis of
large-scale reaction networks. The Journal of Chemical Physics 142:244106).
It has the same functionality support as Wmdirect, but provides significant speedup for suitable models.
steps.API_1.solver.Tetexact implements a stochastic reaction-diffusion
solver, based on Gillespie’s Direct SSA Method extended for diffusive fluxes
between tetrahedral elements in complex geometries.
steps.API_1.solver.TetODE is a spatial determinstic solver where diffusive fluxes are
between tetrahedral elements in complex geometries. Uses CVODE for solutions.
While each unique solver applies a different method for simulation to the other solvers, each solver also typically supports a different subset of all the features in steps.API_1.model and steps.API_1.geom components, where some features are unimplemented because they don’t make sense for the solver or simply because the solver is not yet fully mature. For example, the exclusively well-mixed solvers, Wmdirect and Wmrk4, do not support mesh-based spatial simulations so ignore diffusion rules. When a user attempts to invoke an unimplemented feature on a solver, the feature may simply be ignored (for example a Wmdirect solver object can be created with a mesh-based geometry, but the solver will simply ignore the spatial information and extract only the compartment volumes, patch areas and connectivity) or it may result in a simulation error. Such behaviours are described in more detail in the relevant sections. The following table gives a visual guide to the supported features for each solver:
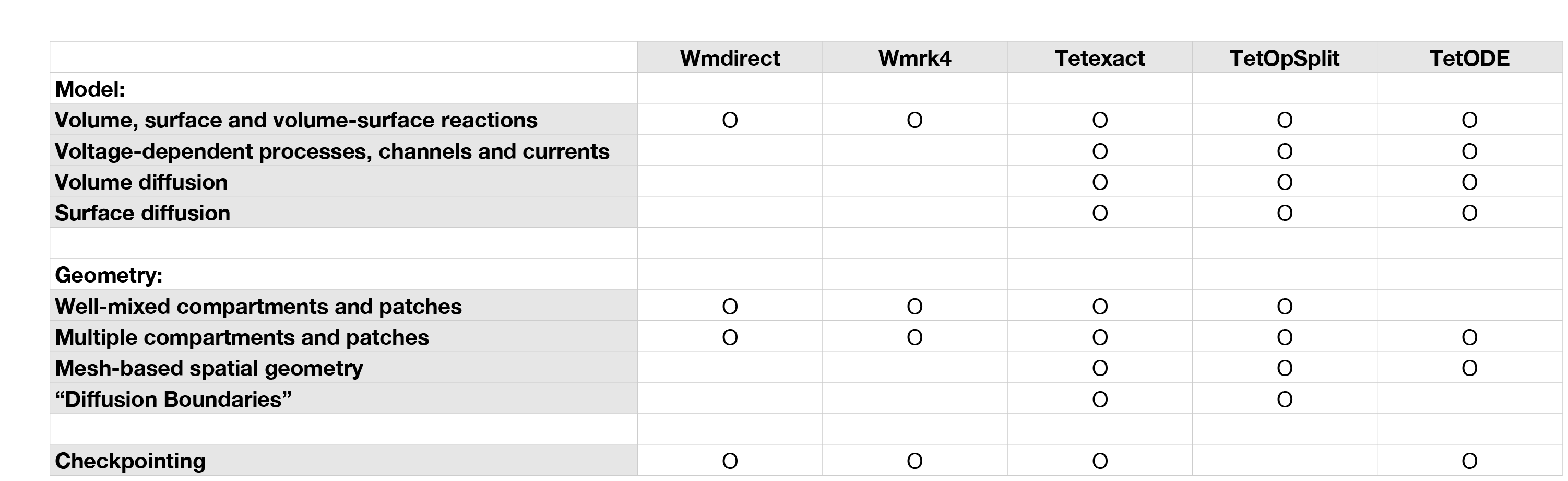
At-a-glance guide of supported features by solver.
- class Wmdirect[source]
Construction:
sim = steps.solver.Wmdirect(model, geom, rng)
Create a non-spatial stochastic solver based on Gillespie’s SSA.
Arguments: steps.model.Model model steps.geom.Geom geom steps.rng.RNG rng
Construction:
sim = steps.solver.Wmdirect(model, geom, rng)
Create a non-spatial stochastic solver based on Gillespie’s SSA.
Arguments: steps.model.Model model steps.geom.Geom geom steps.rng.RNG rng
Solver Information
- getSolverName()
Returns a string of the solver’s name.
Syntax:
getSolverName()
Arguments: None
Return: string
- getSolverDesc()
Returns a string giving a short description of the solver.
Syntax:
getSolverDesc()
Arguments: None
Return: string
- getSolverAuthors()
Returns a string of the solver authors names.
Syntax:
getSolverAuthors()
Arguments: None
Return: string
- getSolverEmail()
Returns a string giving the author’s email address.
Syntax:
getSolverEmail()
Arguments: None
Return: string
Solver Control
- reset()
Reset the simulation to the state the solver was initialised to. Typically, this resets all concentrations of all chemical species in all elements (whether compartments and patches in a well-mixed solver or tetrahedrons and triangles in a mesh-based solver) to zero, resets the simulation time to zero and resets reaction (and diffusion) rates to the default values described in the steps.model objects. All reaction (and diffusion) rules are reset to active and all compartment volumes and patch areas are reset to default values described in steps.geom objects (for well-mixed solvers). Usually, this method should be called before starting each simulation iteration.
Syntax:
reset()
Arguments: None
Return: None
- checkpoint(file_name)
Checkpoint data to a file.
Syntax:
checkpoint(file_name)
Arguments: string file_name
Return: None
- restore(file_name)
Restore data from a file.
Syntax:
restore(file_name)
Arguments: string file_name
Return: None
- run(end_time, cp_interval=0.0, prefix='')[source]
Run the simulation until <end_time>, automatically checkpoint at each <cp_interval>. Prefix can be added using prefix=<prefix_string>.
- advance(advance_time, cp_interval=0.0, prefix='')[source]
Advance the simulation for advance_time, automatically checkpoint at each cp_interval. Prefix can be added using prefix=<prefix_string>.
- step()
Advance the simulation for one ‘step’. In stochastic solvers this is one ‘realization’ of the Gillespie SSA (one reaction ‘event’). In numerical solvers (currently Wmrk4) this is one time-step, with the stepsize defined with the setDT method.
Syntax:
step()
Arguments: None
Return: None
- getTime()
Returns the current simulation time in seconds.
Syntax:
getTime()
Arguments: None
Return: float
- getA0()
Returns the total propensity of the current simulation state (the total propensity multiplied by an infinitesimally small time dt gives the probability that a reaction will occur in that dt). For Tetexact this includes the propensity from the extension of the SSA for diffusive flux between tetrahedral elements in the mesh.
Syntax:
getA0()
Arguments: None
Return: float
- getNSteps()
Return the number of ‘realizations’ of the SSA, the number of reaction (and diffusion) events in stochastic solvers.
Syntax:
getNSteps()
Arguments: None
Return: int
Compartment Data Access
- getCompVol(c)
Returns the volume of compartment with identifier string comp (in m^3).
Syntax:
getCompVol(comp)
Arguments: string comp
Return: float
- setCompVol(c, vol)
Set the volume of compartment with identifier string comp (in m^3).
Syntax:
setCompVol(comp, vol)
Arguments: string comp float vol
Return: None
- getCompCount(c, s)
DEPRECATED Returns the number of molecules of a species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined count from all tetrahedral elements in the compartment.
Syntax:
getCompCount(comp, spec)
Arguments: string comp string spec
Return: float
- setCompCount(c, s, n)
DEPRECATED Set the number of molecules of a species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined count from all tetrahedral elements in the compartment.
Syntax:
setCompCount(comp, spec, nspec)
Arguments: string comp string spec int nspec
Return: None
- getCompAmount(c, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined amount from all tetrahedral elements in the compartment.
Syntax:
getCompAmount(comp, spec)
Arguments: string comp string spec
Return: float
- setCompAmount(c, s, a)
DEPRECATED Set the amount (in mols) of species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined amount from all tetrahedral elements in the compartment.
Syntax:
setCompAmount(comp, spec, amount)
Arguments: string comp string spec float amount
Return: None
- getCompConc(c, s)
DEPRECATED Returns the concentration (in Molar units) of species with identifier string spec in compartment with identifier string comp.
Note: in a mesh-based simulation this is calculated from the combined number of molecules from all tetrahedral elements in the compartment and the total volume of the tetrahedrons.
Syntax:
getCompConc(comp, spec)
Arguments: string comp string spec
Return: float
- setCompConc(c, s, conc)
DEPRECATED Sets the concentration (in Molar units) of species with identifier string spec in compartment with identifier string comp to conc. In a discrete solver the continuous concentration is converted to a discrete number of molecules.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all tetrahedral elements in the compartment (i.e. a uniform distribution).
Syntax:
setCompConc(comp, spec, conc)
Arguments: string comp string spec float conc
Return: None
- getCompClamped(c, s)
DEPRECATED Returns True if species with identifier string spec in compartment with identifier string comp is clamped, which means the concentration remains the same regardless of reactions that consume or produce molecules of this species. Returns False if not.
Note: in a mesh-based simulation it returns True only if the species is clamped in all tetrahedral elements of the compartment.
Syntax:
getCompClamped(comp, spec)
Arguments: string comp string spec
Return: bool
- setCompClamped(c, s, b)
DEPRECATED Sets whether the concentration of species with identifier string spec in compartment with identifier string comp is clamped (clamped = True) or not (clamped = False). If a species is clamped the concentration stays the same regardless of reactions that consume or produce molecules of the species.
Note: in a mesh-based simulation this will set the species to be clamped or not in all tetrahedral elements of the compartment.
Syntax:
setCompClamped(comp, spec, clamped)
Arguments: string comp string spec bool clamped
Return: None
- getCompReacK(c, r)
Returns the macroscopic reaction constant of reaction with identifier string reac in compartment with identifier string comp. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation the value for the compartment is returned, although individual tetrahedral elements may have different values (set with setTetReacK).
Syntax:
getCompReacK(comp, reac)
Arguments: string comp string reac
Return: float
- setCompReacK(c, r, kf)
Sets the macroscopic reaction constant of reaction with identifier string reac in compartment with identifier string comp to kf. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation this method sets the reaction constant in all tetrahedral elements of the compartment to kf
Note: The default value still comes from the steps.model description, so calling reset() will return the reaction constant to that value.
Syntax:
setCompReacK(comp, reac, kf)
Arguments: string comp string reac float kf
Return: None
- getCompReacActive(c, r)
Returns whether a reaction with identifier string reac in compartment with identifier string comp is active (True) or not (False). If it’s not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this method will return True only if the reaction is active in all tetrahedral elements in the compartment.
Syntax:
getCompReacActive(comp, reac)
Arguments: string comp string reac
Return: bool
- setCompReacActive(c, r, a)
Activate (active = True) or deactivate (active = False) a reaction with identifier string reac in compartment with identifier string comp. If a reaction is not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this will activate/deactivate the reaction in all tetrahedral elements in the compartment.
Syntax:
setCompReacActive(comp, reac, active)
Arguments: string comp string reac bool active
Return: None
- getCompReacC(c, r)
Returns the ‘stochastic reaction constant’ (or ‘specific probability rate constant’) of reaction with identifier string reac in compartment with identifier string comp.
The ‘stochastic reaction constant’ multiplied by infinitesimal time interval dt gives the average probability that one reaction channel of this reaction type will react accordingly in dt.
Note: in a mesh-based simulation (i.e. Tetexact), the stochastic reaction constant is computed as the weighted mean of the stochastic reaction constants in all tetrahedral elements of the compartment.
Syntax:
getCompReacC(comp, reac)
Arguments: string comp string reac
Return: float
- getCompReacH(c, r)
Returns h_mu, the distinct number of ways in which reaction with identifier string reac can occur in compartment with identifier string comp, by computing the product of its reactants. Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the h_mu’s over all tetrahedral elements in the compartment.
Syntax:
getCompReacH(comp, reac)
Arguments: string comp string reac
Return: float
- getCompReacA(c, r)
Returns the propensity of reaction with identifier string reac in compartment with identifier string comp.
The propensity of a reaction is a function of state and is defined as the function whose product with infinitesimal time dt gives the probability that the reaction will occur in the next dt. It is the ‘stochastic reaction constant’ multiplied by ‘h_mu’.
Note: in a mesh-based simulation (i.e. Tetexact), the propensity of a reaction in a compartment is computed as the sum of the propensities in all tetrahedral elements of the compartment.
Syntax:
getCompReacA(comp, reac)
Arguments: string comp string reac
Return: float
- getCompReacExtent(c, r)
Return the extent of reaction with identifier string reac in compartment with identifier string comp, that is the number of times the reaction has occurred up to the current simulation time.
Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the reaction extents in all tetrahedral elements of the compartment.
Syntax:
getCompReacExtent(comp, reac)
Arguments: string comp string reac
Return: index_t
- resetCompReacExtent(c, r)
Resets the extent of reaction with identifier string reac in compartment with identifier string comp to zero.
Note: in a mesh-based simulation (i.e. Tetexact), resets the extents of the reaction in all tetrahedral elements of the compartment.
Syntax:
resetCompReacExtent(comp, reac)
Arguments: string comp string reac
Return: None
Patch Data Access
- getPatchArea(p)
Returns the area of patch with identifier string patch (in m^2).
Syntax:
getPatchArea(patch)
Arguments: string patch
Return: float
- setPatchArea(p, area)
Sets the area of patch with identifier string patch to area a (in m^2).
Syntax:
setPatchArea(patch, area)
Arguments: string patch float area
Return: None
- getPatchCount(p, s)
DEPRECATED Returns the number of molecules of species with identifier string spec in patch with identifier string patch.Note: in a mesh-based simulation this is the combined count from all triangular elements in the patch.
Syntax:
getPatchCount(patch, spec)
Arguments: string patch string spec
Return: float
- setPatchCount(p, s, n)
DEPRECATED Sets the number of molecules of species with identifier string spec in patch with identifier string patch to n. Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
setPatchCount(patch, spec, n)
Arguments: string patch string spec int n
Return: float
- getPatchAmount(p, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in patch with identifier string patch.
Note: in a mesh-based simulation this is the combined amount from all triangular elements in the patch.
Syntax:
getPatchAmount(patch, spec)
Arguments: string patch string spec
Return: float
- setPatchAmount(p, s, a)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in patch with identifier string patch to a. In a discrete solver, such as Wmdirect and Tetexact, this continuous value is converted internally into a discrete number of molecules by multiplication with Avogadro’s number.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
setPatchAmount(patch, spec, a)
Arguments: string patch string spec float a
Return: None
- getPatchClamped(p, s)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in patch with identifier string patch to a. In a discrete solver, such as Wmdirect and Tetexact, this continuous value is converted internally into a discrete number of molecules by multiplication with Avogadro’s number.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
getPatchClamped(patch, spec)
Arguments: string patch string spec
Return: bool
- setPatchClamped(p, s, buf)
DEPRECATED Sets whether the species with identifier string spec in patch with identifier string patch is clamped (clamped = True) or not (clamped = False). If a species is clamped the number of molecules stays the same regardless of surface reactions that consume or produce molecules of the species.
Note: in a mesh-based simulation this will set the species to be clamped in all triangular elements of the patch.
Syntax:
setPatchClamped(patch, spec, clamped)
Arguments: string patch string spec bool clamped
Return: None
- getPatchSReacK(p, r)
Returns the macroscopic reaction constant of surface reaction with identifier string sreac in patch with identifier string patch. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based solver the value for the patch is returned, although individual triangle elements may have different values (set with setTriSReacK).
Syntax:
getPatchSReacK(patch, reac)
Arguments: string patch string reac
Return: float
- setPatchSReacK(p, r, kf)
Sets the macroscopic reaction constant of surface reaction with identifier string sreac in patch with identifier string patch to kf. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation this method sets the surface reaction constant in all triangular elements of the patch to kf.
Note: The default value still comes from the steps.model description, so calling reset() will return the surface reaction constant to that value.
Syntax:
setPatchSReacK(patch, reac, kf)
Arguments: string patch string reac float kf
Return: None
- getPatchSReacActive(p, r)
Returns whether a surface reaction with identifier string sreac in patch with identifier string patch is active (True) or not (False). If it’s not active this means that a surface reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this method will return True only if the surface reaction is active in all triangular elements in the patch.
Syntax:
getPatchSReacActive(patch, reac)
Arguments: string patch string reac
Return: bool
- setPatchSReacActive(p, r, a)
Activate (active = True) or deactivate (active = False) a surface reaction with identifier string sreac in patch with identifier string patch. If a surface reaction is not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this will activate/ deactivate the reaction in all triangular elements in the patch.
Syntax:
setPatchSReacActive(patch, reac, active)
Arguments: string patch string reac bool active
Return: None
- getPatchSReacC(p, r)
Returns the ‘stochastic reaction constant’ (or ‘specific probability rate constant’) of surface reaction with identifier string sreac in patch with identifier string patch.
Note: in a mesh-based simulation (i.e. Tetexact), the stochastic reaction constant is computed as the weighted mean of the stochastic reaction constants in all triangular elements of the patch.
Syntax:
getPatchSReacC(patch, reac)
Arguments: string patch string reac
Return: float
- getPatchSReacH(p, r)
Returns h_mu, the distinct number of ways in which surface reaction with identifier string sreac can occur in patch with identifier string patch, by computing the product of its reactants. Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the h_mu’s over all triangular elements in the patch.
Syntax:
getPatchSReacH(patch, reac)
Arguments: string patch string reac
Return: float
- getPatchSReacA(p, r)
Returns the propensity of surface reaction with identifier string sreac in patch with identifier string patch. Note: in a mesh-based simulation (i.e. Tetexact), the propensity of a surface reaction in a patch is computed as the sum of the propensities in all triangular elements of the patch.
Syntax:
getPatchSReacA(patch, reac)
Arguments: string patch string reac
Return: float
- getPatchSReacExtent(p, r)
Returns the extent of surface reaction with identifier string sreac in patch with identifier string patch, that is the number of times the surface reaction has occurred up to the current simulation time.
Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the reaction extents in all triangular elements of the patch.
Syntax:
getPatchSReacExtent(patch, reac)
Arguments: string patch string reac
Return: index_t
- resetPatchSReacExtent(p, r)
Resets the extent of reaction with identifier string sreac in patch with identifier string patch to zero.
Note: in a mesh-based simulation (i.e. Tetexact), resets the extents of the reaction in all triangular elements of the patch.
Syntax:
resetPatchSReacExtent(patch, reac)
Arguments: string patch string reac
Return: None
- class Wmrk4[source]
Construction:
sim = steps.solver.Wmrk4(model, geom)
Create a non-spatial deterministic solver based on the Runge-Kutta fourth order method.
Arguments: steps.model.Model model steps.geom.Geom geom
Construction:
sim = steps.solver.Wmrk4(model, geom)
Create a non-spatial deterministic solver based on the Runge-Kutta fourth order method.
Arguments: steps.model.Model model steps.geom.Geom geom
Solver Information
- getSolverName()
Returns a string of the solver’s name.
Syntax:
getSolverName()
Arguments: None
Return: string
- getSolverDesc()
Returns a string giving a short description of the solver.
Syntax:
getSolverDesc()
Arguments: None
Return: string
- getSolverAuthors()
Returns a string of the solver authors names.
Syntax:
getSolverAuthors()
Arguments: None
Return: string
- getSolverEmail()
Returns a string giving the author’s email address.
Syntax:
getSolverEmail()
Arguments: None
Return: string
Solver Control
- reset()
Reset the simulation to the state the solver was initialised to. Typically, this resets all concentrations of all chemical species in all elements (whether compartments and patches in a well-mixed solver or tetrahedrons and triangles in a mesh-based solver) to zero, resets the simulation time to zero and resets reaction (and diffusion) rates to the default values described in the steps.model objects. All reaction (and diffusion) rules are reset to active and all compartment volumes and patch areas are reset to default values described in steps.geom objects (for well-mixed solvers). Usually, this method should be called before starting each simulation iteration.
Syntax:
reset()
Arguments: None
Return: None
- checkpoint(file_name)
Checkpoint data to a file.
Syntax:
checkpoint(file_name)
Arguments: string file_name
Return: None
- restore(file_name)
Restore data from a file.
Syntax:
restore(file_name)
Arguments: string file_name
Return: None
- run(end_time, cp_interval=0.0, prefix='')[source]
Run the simulation until end_time, automatically checkpoint at each cp_interval. Prefix can be added using prefix=<prefix_string>.
- advance(advance_time, cp_interval=0.0, prefix='')[source]
Advance the simulation for advance_time, automatically checkpoint at each cp_interval. Prefix can be added using prefix=<prefix_string>.
- step()
Advance the simulation for one ‘step’. In stochastic solvers this is one ‘realization’ of the Gillespie SSA (one reaction ‘event’). In numerical solvers (currently Wmrk4) this is one time-step, with the stepsize defined with the setDT method.
Syntax:
step()
Arguments: None
Return: None
- setRk4DT(dt)
Set the stepsize for numerical solvers. Must be called before running a simulation with these solvers (currently Wmrk4) since there is no default stepsize. The deterministic solver Wmrk4 implements a fixed stepsize (i.e. not adaptive), although the stepsize can be altered at any point during the simulation with this method.
Syntax:
setRk4tDT(dt)
Arguments: float dt
Return: None
- getTime()
Returns the current simulation time in seconds.
Syntax:
getTime()
Arguments: None
Return: float
Compartment Data Access
- getCompVol(c)
Returns the volume of compartment with identifier string comp (in m^3).
Syntax:
getCompVol(comp)
Arguments: string comp
Return: float
- setCompVol(c, vol)
Set the volume of compartment with identifier string comp (in m^3).
Syntax:
setCompVol(comp, vol)
Arguments: string comp float vol
Return: None
- getCompCount(c, s)
DEPRECATED Returns the number of molecules of a species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined count from all tetrahedral elements in the compartment.
Syntax:
getCompCount(comp, spec)
Arguments: string comp string spec
Return: float
- setCompCount(c, s, n)
DEPRECATED Set the number of molecules of a species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined count from all tetrahedral elements in the compartment.
Syntax:
setCompCount(comp, spec, nspec)
Arguments: string comp string spec int nspec
Return: None
- getCompAmount(c, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined amount from all tetrahedral elements in the compartment.
Syntax:
getCompAmount(comp, spec)
Arguments: string comp string spec
Return: float
- setCompAmount(c, s, a)
DEPRECATED Set the amount (in mols) of species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined amount from all tetrahedral elements in the compartment.
Syntax:
setCompAmount(comp, spec, amount)
Arguments: string comp string spec float amount
Return: None
- getCompConc(c, s)
DEPRECATED Returns the concentration (in Molar units) of species with identifier string spec in compartment with identifier string comp.
Note: in a mesh-based simulation this is calculated from the combined number of molecules from all tetrahedral elements in the compartment and the total volume of the tetrahedrons.
Syntax:
getCompConc(comp, spec)
Arguments: string comp string spec
Return: float
- setCompConc(c, s, conc)
DEPRECATED Sets the concentration (in Molar units) of species with identifier string spec in compartment with identifier string comp to conc. In a discrete solver the continuous concentration is converted to a discrete number of molecules.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all tetrahedral elements in the compartment (i.e. a uniform distribution).
Syntax:
setCompConc(comp, spec, conc)
Arguments: string comp string spec float conc
Return: None
- getCompClamped(c, s)
DEPRECATED Returns True if species with identifier string spec in compartment with identifier string comp is clamped, which means the concentration remains the same regardless of reactions that consume or produce molecules of this species. Returns False if not.
Note: in a mesh-based simulation it returns True only if the species is clamped in all tetrahedral elements of the compartment.
Syntax:
getCompClamped(comp, spec)
Arguments: string comp string spec
Return: bool
- setCompClamped(c, s, b)
DEPRECATED Sets whether the concentration of species with identifier string spec in compartment with identifier string comp is clamped (clamped = True) or not (clamped = False). If a species is clamped the concentration stays the same regardless of reactions that consume or produce molecules of the species.
Note: in a mesh-based simulation this will set the species to be clamped or not in all tetrahedral elements of the compartment.
Syntax:
setCompClamped(comp, spec, clamped)
Arguments: string comp string spec bool clamped
Return: None
- getCompReacK(c, r)
Returns the macroscopic reaction constant of reaction with identifier string reac in compartment with identifier string comp. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation the value for the compartment is returned, although individual tetrahedral elements may have different values (set with setTetReacK).
Syntax:
getCompReacK(comp, reac)
Arguments: string comp string reac
Return: float
- setCompReacK(c, r, kf)
Sets the macroscopic reaction constant of reaction with identifier string reac in compartment with identifier string comp to kf. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation this method sets the reaction constant in all tetrahedral elements of the compartment to kf
Note: The default value still comes from the steps.model description, so calling reset() will return the reaction constant to that value.
Syntax:
setCompReacK(comp, reac, kf)
Arguments: string comp string reac float kf
Return: None
- getCompReacActive(c, r)
Returns whether a reaction with identifier string reac in compartment with identifier string comp is active (True) or not (False). If it’s not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this method will return True only if the reaction is active in all tetrahedral elements in the compartment.
Syntax:
getCompReacActive(comp, reac)
Arguments: string comp string reac
Return: bool
- setCompReacActive(c, r, a)
Activate (active = True) or deactivate (active = False) a reaction with identifier string reac in compartment with identifier string comp. If a reaction is not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this will activate/deactivate the reaction in all tetrahedral elements in the compartment.
Syntax:
setCompReacActive(comp, reac, active)
Arguments: string comp string reac bool active
Return: None
Patch Data Access
- getPatchArea(p)
Returns the area of patch with identifier string patch (in m^2).
Syntax:
getPatchArea(patch)
Arguments: string patch
Return: float
- setPatchArea(p, area)
Sets the area of patch with identifier string patch to area a (in m^2).
Syntax:
setPatchArea(patch, area)
Arguments: string patch float area
Return: None
- getPatchCount(p, s)
DEPRECATED Returns the number of molecules of species with identifier string spec in patch with identifier string patch.Note: in a mesh-based simulation this is the combined count from all triangular elements in the patch.
Syntax:
getPatchCount(patch, spec)
Arguments: string patch string spec
Return: float
- setPatchCount(p, s, n)
DEPRECATED Sets the number of molecules of species with identifier string spec in patch with identifier string patch to n. Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
setPatchCount(patch, spec, n)
Arguments: string patch string spec int n
Return: float
- getPatchAmount(p, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in patch with identifier string patch.
Note: in a mesh-based simulation this is the combined amount from all triangular elements in the patch.
Syntax:
getPatchAmount(patch, spec)
Arguments: string patch string spec
Return: float
- setPatchAmount(p, s, a)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in patch with identifier string patch to a. In a discrete solver, such as Wmdirect and Tetexact, this continuous value is converted internally into a discrete number of molecules by multiplication with Avogadro’s number.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
setPatchAmount(patch, spec, a)
Arguments: string patch string spec float a
Return: None
- getPatchClamped(p, s)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in patch with identifier string patch to a. In a discrete solver, such as Wmdirect and Tetexact, this continuous value is converted internally into a discrete number of molecules by multiplication with Avogadro’s number.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
getPatchClamped(patch, spec)
Arguments: string patch string spec
Return: bool
- setPatchClamped(p, s, buf)
DEPRECATED Sets whether the species with identifier string spec in patch with identifier string patch is clamped (clamped = True) or not (clamped = False). If a species is clamped the number of molecules stays the same regardless of surface reactions that consume or produce molecules of the species.
Note: in a mesh-based simulation this will set the species to be clamped in all triangular elements of the patch.
Syntax:
setPatchClamped(patch, spec, clamped)
Arguments: string patch string spec bool clamped
Return: None
- getPatchSReacK(p, r)
Returns the macroscopic reaction constant of surface reaction with identifier string sreac in patch with identifier string patch. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based solver the value for the patch is returned, although individual triangle elements may have different values (set with setTriSReacK).
Syntax:
getPatchSReacK(patch, reac)
Arguments: string patch string reac
Return: float
- setPatchSReacK(p, r, kf)
Sets the macroscopic reaction constant of surface reaction with identifier string sreac in patch with identifier string patch to kf. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation this method sets the surface reaction constant in all triangular elements of the patch to kf.
Note: The default value still comes from the steps.model description, so calling reset() will return the surface reaction constant to that value.
Syntax:
setPatchSReacK(patch, reac, kf)
Arguments: string patch string reac float kf
Return: None
- getPatchSReacActive(p, r)
Returns whether a surface reaction with identifier string sreac in patch with identifier string patch is active (True) or not (False). If it’s not active this means that a surface reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this method will return True only if the surface reaction is active in all triangular elements in the patch.
Syntax:
getPatchSReacActive(patch, reac)
Arguments: string patch string reac
Return: bool
- setPatchSReacActive(p, r, a)
Activate (active = True) or deactivate (active = False) a surface reaction with identifier string sreac in patch with identifier string patch. If a surface reaction is not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this will activate/ deactivate the reaction in all triangular elements in the patch.
Syntax:
setPatchSReacActive(patch, reac, active)
Arguments: string patch string reac bool active
Return: None
- class Wmrssa[source]
Construction:
sim = steps.solver.Wmrssa(model, geom, rng)
Create a non-spatial stochastic solver implementing a rejection-based SSA.
Arguments: steps.model.Model model steps.geom.Geom geom steps.rng.RNG rng
Construction:
sim = steps.solver.Wmrssa(model, geom, rng)
Create a non-spatial stochastic solver based on Gillespie’s SSA.
Arguments: steps.model.Model model steps.geom.Geom geom steps.rng.RNG rng
Solver Information
- getSolverName()
Returns a string of the solver’s name.
Syntax:
getSolverName()
Arguments: None
Return: string
- getSolverDesc()
Returns a string giving a short description of the solver.
Syntax:
getSolverDesc()
Arguments: None
Return: string
- getSolverAuthors()
Returns a string of the solver authors names.
Syntax:
getSolverAuthors()
Arguments: None
Return: string
- getSolverEmail()
Returns a string giving the author’s email address.
Syntax:
getSolverEmail()
Arguments: None
Return: string
Solver Control
- reset()
Reset the simulation to the state the solver was initialised to. Typically, this resets all concentrations of all chemical species in all elements (whether compartments and patches in a well-mixed solver or tetrahedrons and triangles in a mesh-based solver) to zero, resets the simulation time to zero and resets reaction (and diffusion) rates to the default values described in the steps.model objects. All reaction (and diffusion) rules are reset to active and all compartment volumes and patch areas are reset to default values described in steps.geom objects (for well-mixed solvers). Usually, this method should be called before starting each simulation iteration.
Syntax:
reset()
Arguments: None
Return: None
- checkpoint(file_name)
Checkpoint data to a file.
Syntax:
checkpoint(file_name)
Arguments: string file_name
Return: None
- restore(file_name)
Restore data from a file.
Syntax:
restore(file_name)
Arguments: string file_name
Return: None
- run(end_time, cp_interval=0.0, prefix='')[source]
Run the simulation until <end_time>, automatically checkpoint at each <cp_interval>. Prefix can be added using prefix=<prefix_string>.
- advance(advance_time, cp_interval=0.0, prefix='')[source]
Advance the simulation for advance_time, automatically checkpoint at each cp_interval. Prefix can be added using prefix=<prefix_string>.
- step()
Advance the simulation for one ‘step’. In stochastic solvers this is one ‘realization’ of the Gillespie SSA (one reaction ‘event’). In numerical solvers (currently Wmrk4) this is one time-step, with the stepsize defined with the setDT method.
Syntax:
step()
Arguments: None
Return: None
- getTime()
Returns the current simulation time in seconds.
Syntax:
getTime()
Arguments: None
Return: float
- getNSteps()
Return the number of ‘realizations’ of the SSA, the number of reaction (and diffusion) events in stochastic solvers.
Syntax:
getNSteps()
Arguments: None
Return: int
Compartment Data Access
- getCompVol(c)
Returns the volume of compartment with identifier string comp (in m^3).
Syntax:
getCompVol(comp)
Arguments: string comp
Return: float
- setCompVol(c, vol)
Set the volume of compartment with identifier string comp (in m^3).
Syntax:
setCompVol(comp, vol)
Arguments: string comp float vol
Return: None
- getCompCount(c, s)
DEPRECATED Returns the number of molecules of a species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined count from all tetrahedral elements in the compartment.
Syntax:
getCompCount(comp, spec)
Arguments: string comp string spec
Return: float
- setCompCount(c, s, n)
DEPRECATED Set the number of molecules of a species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined count from all tetrahedral elements in the compartment.
Syntax:
setCompCount(comp, spec, nspec)
Arguments: string comp string spec int nspec
Return: None
- getCompAmount(c, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined amount from all tetrahedral elements in the compartment.
Syntax:
getCompAmount(comp, spec)
Arguments: string comp string spec
Return: float
- setCompAmount(c, s, a)
DEPRECATED Set the amount (in mols) of species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined amount from all tetrahedral elements in the compartment.
Syntax:
setCompAmount(comp, spec, amount)
Arguments: string comp string spec float amount
Return: None
- getCompConc(c, s)
DEPRECATED Returns the concentration (in Molar units) of species with identifier string spec in compartment with identifier string comp.
Note: in a mesh-based simulation this is calculated from the combined number of molecules from all tetrahedral elements in the compartment and the total volume of the tetrahedrons.
Syntax:
getCompConc(comp, spec)
Arguments: string comp string spec
Return: float
- setCompConc(c, s, conc)
DEPRECATED Sets the concentration (in Molar units) of species with identifier string spec in compartment with identifier string comp to conc. In a discrete solver the continuous concentration is converted to a discrete number of molecules.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all tetrahedral elements in the compartment (i.e. a uniform distribution).
Syntax:
setCompConc(comp, spec, conc)
Arguments: string comp string spec float conc
Return: None
- getCompClamped(c, s)
DEPRECATED Returns True if species with identifier string spec in compartment with identifier string comp is clamped, which means the concentration remains the same regardless of reactions that consume or produce molecules of this species. Returns False if not.
Note: in a mesh-based simulation it returns True only if the species is clamped in all tetrahedral elements of the compartment.
Syntax:
getCompClamped(comp, spec)
Arguments: string comp string spec
Return: bool
- setCompClamped(c, s, b)
DEPRECATED Sets whether the concentration of species with identifier string spec in compartment with identifier string comp is clamped (clamped = True) or not (clamped = False). If a species is clamped the concentration stays the same regardless of reactions that consume or produce molecules of the species.
Note: in a mesh-based simulation this will set the species to be clamped or not in all tetrahedral elements of the compartment.
Syntax:
setCompClamped(comp, spec, clamped)
Arguments: string comp string spec bool clamped
Return: None
- getCompReacK(c, r)
Returns the macroscopic reaction constant of reaction with identifier string reac in compartment with identifier string comp. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation the value for the compartment is returned, although individual tetrahedral elements may have different values (set with setTetReacK).
Syntax:
getCompReacK(comp, reac)
Arguments: string comp string reac
Return: float
- setCompReacK(c, r, kf)
Sets the macroscopic reaction constant of reaction with identifier string reac in compartment with identifier string comp to kf. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation this method sets the reaction constant in all tetrahedral elements of the compartment to kf
Note: The default value still comes from the steps.model description, so calling reset() will return the reaction constant to that value.
Syntax:
setCompReacK(comp, reac, kf)
Arguments: string comp string reac float kf
Return: None
- getCompReacActive(c, r)
Returns whether a reaction with identifier string reac in compartment with identifier string comp is active (True) or not (False). If it’s not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this method will return True only if the reaction is active in all tetrahedral elements in the compartment.
Syntax:
getCompReacActive(comp, reac)
Arguments: string comp string reac
Return: bool
- setCompReacActive(c, r, a)
Activate (active = True) or deactivate (active = False) a reaction with identifier string reac in compartment with identifier string comp. If a reaction is not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this will activate/deactivate the reaction in all tetrahedral elements in the compartment.
Syntax:
setCompReacActive(comp, reac, active)
Arguments: string comp string reac bool active
Return: None
- getCompReacC(c, r)
Returns the ‘stochastic reaction constant’ (or ‘specific probability rate constant’) of reaction with identifier string reac in compartment with identifier string comp.
The ‘stochastic reaction constant’ multiplied by infinitesimal time interval dt gives the average probability that one reaction channel of this reaction type will react accordingly in dt.
Note: in a mesh-based simulation (i.e. Tetexact), the stochastic reaction constant is computed as the weighted mean of the stochastic reaction constants in all tetrahedral elements of the compartment.
Syntax:
getCompReacC(comp, reac)
Arguments: string comp string reac
Return: float
- getCompReacH(c, r)
Returns h_mu, the distinct number of ways in which reaction with identifier string reac can occur in compartment with identifier string comp, by computing the product of its reactants. Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the h_mu’s over all tetrahedral elements in the compartment.
Syntax:
getCompReacH(comp, reac)
Arguments: string comp string reac
Return: float
- getCompReacA(c, r)
Returns the propensity of reaction with identifier string reac in compartment with identifier string comp.
The propensity of a reaction is a function of state and is defined as the function whose product with infinitesimal time dt gives the probability that the reaction will occur in the next dt. It is the ‘stochastic reaction constant’ multiplied by ‘h_mu’.
Note: in a mesh-based simulation (i.e. Tetexact), the propensity of a reaction in a compartment is computed as the sum of the propensities in all tetrahedral elements of the compartment.
Syntax:
getCompReacA(comp, reac)
Arguments: string comp string reac
Return: float
- getCompReacExtent(c, r)
Return the extent of reaction with identifier string reac in compartment with identifier string comp, that is the number of times the reaction has occurred up to the current simulation time.
Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the reaction extents in all tetrahedral elements of the compartment.
Syntax:
getCompReacExtent(comp, reac)
Arguments: string comp string reac
Return: index_t
- resetCompReacExtent(c, r)
Resets the extent of reaction with identifier string reac in compartment with identifier string comp to zero.
Note: in a mesh-based simulation (i.e. Tetexact), resets the extents of the reaction in all tetrahedral elements of the compartment.
Syntax:
resetCompReacExtent(comp, reac)
Arguments: string comp string reac
Return: None
Patch Data Access
- getPatchArea(p)
Returns the area of patch with identifier string patch (in m^2).
Syntax:
getPatchArea(patch)
Arguments: string patch
Return: float
- setPatchArea(p, area)
Sets the area of patch with identifier string patch to area a (in m^2).
Syntax:
setPatchArea(patch, area)
Arguments: string patch float area
Return: None
- getPatchCount(p, s)
DEPRECATED Returns the number of molecules of species with identifier string spec in patch with identifier string patch.Note: in a mesh-based simulation this is the combined count from all triangular elements in the patch.
Syntax:
getPatchCount(patch, spec)
Arguments: string patch string spec
Return: float
- setPatchCount(p, s, n)
DEPRECATED Sets the number of molecules of species with identifier string spec in patch with identifier string patch to n. Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
setPatchCount(patch, spec, n)
Arguments: string patch string spec int n
Return: float
- getPatchAmount(p, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in patch with identifier string patch.
Note: in a mesh-based simulation this is the combined amount from all triangular elements in the patch.
Syntax:
getPatchAmount(patch, spec)
Arguments: string patch string spec
Return: float
- setPatchAmount(p, s, a)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in patch with identifier string patch to a. In a discrete solver, such as Wmdirect and Tetexact, this continuous value is converted internally into a discrete number of molecules by multiplication with Avogadro’s number.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
setPatchAmount(patch, spec, a)
Arguments: string patch string spec float a
Return: None
- getPatchClamped(p, s)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in patch with identifier string patch to a. In a discrete solver, such as Wmdirect and Tetexact, this continuous value is converted internally into a discrete number of molecules by multiplication with Avogadro’s number.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
getPatchClamped(patch, spec)
Arguments: string patch string spec
Return: bool
- setPatchClamped(p, s, buf)
DEPRECATED Sets whether the species with identifier string spec in patch with identifier string patch is clamped (clamped = True) or not (clamped = False). If a species is clamped the number of molecules stays the same regardless of surface reactions that consume or produce molecules of the species.
Note: in a mesh-based simulation this will set the species to be clamped in all triangular elements of the patch.
Syntax:
setPatchClamped(patch, spec, clamped)
Arguments: string patch string spec bool clamped
Return: None
- getPatchSReacK(p, r)
Returns the macroscopic reaction constant of surface reaction with identifier string sreac in patch with identifier string patch. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based solver the value for the patch is returned, although individual triangle elements may have different values (set with setTriSReacK).
Syntax:
getPatchSReacK(patch, reac)
Arguments: string patch string reac
Return: float
- setPatchSReacK(p, r, kf)
Sets the macroscopic reaction constant of surface reaction with identifier string sreac in patch with identifier string patch to kf. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation this method sets the surface reaction constant in all triangular elements of the patch to kf.
Note: The default value still comes from the steps.model description, so calling reset() will return the surface reaction constant to that value.
Syntax:
setPatchSReacK(patch, reac, kf)
Arguments: string patch string reac float kf
Return: None
- getPatchSReacActive(p, r)
Returns whether a surface reaction with identifier string sreac in patch with identifier string patch is active (True) or not (False). If it’s not active this means that a surface reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this method will return True only if the surface reaction is active in all triangular elements in the patch.
Syntax:
getPatchSReacActive(patch, reac)
Arguments: string patch string reac
Return: bool
- setPatchSReacActive(p, r, a)
Activate (active = True) or deactivate (active = False) a surface reaction with identifier string sreac in patch with identifier string patch. If a surface reaction is not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this will activate/ deactivate the reaction in all triangular elements in the patch.
Syntax:
setPatchSReacActive(patch, reac, active)
Arguments: string patch string reac bool active
Return: None
- getPatchSReacC(p, r)
Returns the ‘stochastic reaction constant’ (or ‘specific probability rate constant’) of surface reaction with identifier string sreac in patch with identifier string patch.
Note: in a mesh-based simulation (i.e. Tetexact), the stochastic reaction constant is computed as the weighted mean of the stochastic reaction constants in all triangular elements of the patch.
Syntax:
getPatchSReacC(patch, reac)
Arguments: string patch string reac
Return: float
- getPatchSReacH(p, r)
Returns h_mu, the distinct number of ways in which surface reaction with identifier string sreac can occur in patch with identifier string patch, by computing the product of its reactants. Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the h_mu’s over all triangular elements in the patch.
Syntax:
getPatchSReacH(patch, reac)
Arguments: string patch string reac
Return: float
- getPatchSReacA(p, r)
Returns the propensity of surface reaction with identifier string sreac in patch with identifier string patch. Note: in a mesh-based simulation (i.e. Tetexact), the propensity of a surface reaction in a patch is computed as the sum of the propensities in all triangular elements of the patch.
Syntax:
getPatchSReacA(patch, reac)
Arguments: string patch string reac
Return: float
- getPatchSReacExtent(p, r)
Returns the extent of surface reaction with identifier string sreac in patch with identifier string patch, that is the number of times the surface reaction has occurred up to the current simulation time.
Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the reaction extents in all triangular elements of the patch.
Syntax:
getPatchSReacExtent(patch, reac)
Arguments: string patch string reac
Return: index_t
- resetPatchSReacExtent(p, r)
Resets the extent of reaction with identifier string sreac in patch with identifier string patch to zero.
Note: in a mesh-based simulation (i.e. Tetexact), resets the extents of the reaction in all triangular elements of the patch.
Syntax:
resetPatchSReacExtent(patch, reac)
Arguments: string patch string reac
Return: None
- class Tetexact[source]
Construction:
sim = steps.solver.Tetexact(model, geom, rng, calcMembPot = 0)
Create a spatial stochastic solver based on Gillespie’s SSA, extended with diffusion across elements in a tetrahedral mesh. If voltage is to be simulated, argument calcMemPot=1 will set to the default solver. calcMembPot=0 means voltage will not be simulated.
Arguments: steps.model.Model model steps.geom.Geom geom steps.rng.RNG rng int calcMemPot (default=0)
Construction:
sim = steps.solver.Tetexact(model, geom, rng, calcMembPot = 0, calcMembPot_lenient=False)
Create a spatial stochastic solver based on Gillespie’s SSA, extended with diffusion across elements in a tetrahedral mesh. If voltage is to be simulated, argument calcMemPot=1 will set to the default solver. calcMembPot=0 means voltage will not be simulated.
Arguments: steps.model.Model model steps.geom.Geom geom steps.rng.RNG rng int calcMemPot (default=0) bool calcMembPot_lenient (default=False)
Solver Information
- getSolverName()
Returns a string of the solver’s name.
Syntax:
getSolverName()
Arguments: None
Return: string
- getSolverDesc()
Returns a string giving a short description of the solver.
Syntax:
getSolverDesc()
Arguments: None
Return: string
- getSolverAuthors()
Returns a string of the solver authors names.
Syntax:
getSolverAuthors()
Arguments: None
Return: string
- getSolverEmail()
Returns a string giving the author’s email address.
Syntax:
getSolverEmail()
Arguments: None
Return: string
Solver Control
- reset()
Reset the simulation to the state the solver was initialised to. Typically, this resets all concentrations of all chemical species in all elements (whether compartments and patches in a well-mixed solver or tetrahedrons and triangles in a mesh-based solver) to zero, resets the simulation time to zero and resets reaction (and diffusion) rates to the default values described in the steps.model objects. All reaction (and diffusion) rules are reset to active and all compartment volumes and patch areas are reset to default values described in steps.geom objects (for well-mixed solvers). Usually, this method should be called before starting each simulation iteration.
Syntax:
reset()
Arguments: None
Return: None
- checkpoint(file_name)
Checkpoint data to a file.
Syntax:
checkpoint(file_name)
Arguments: string file_name
Return: None
- restore(file_name)
Restore data from a file.
Syntax:
restore(file_name)
Arguments: string file_name
Return: None
- run(end_time, cp_interval=0.0, prefix='')[source]
Run the simulation until <end_time>, automatically checkpoint at each <cp_interval>. Prefix can be added using prefix=<prefix_string>.
- advance(advance_time, cp_interval=0.0, prefix='')[source]
Advance the simulation for <advance_time>, automatically checkpoint at each <cp_interval>. Prefix can be added using prefix=<prefix_string>.
- step()
Advance the simulation for one ‘step’. In stochastic solvers this is one ‘realization’ of the Gillespie SSA (one reaction ‘event’). In numerical solvers (currently Wmrk4) this is one time-step, with the stepsize defined with the setDT method.
Syntax:
step()
Arguments: None
Return: None
- getTime()
Returns the current simulation time in seconds.
Syntax:
getTime()
Arguments: None
Return: float
- getA0()
Returns the total propensity of the current simulation state (the total propensity multiplied by an infinitesimally small time dt gives the probability that a reaction will occur in that dt). For Tetexact this includes the propensity from the extension of the SSA for diffusive flux between tetrahedral elements in the mesh.
Syntax:
getA0()
Arguments: None
Return: float
- getNSteps()
Return the number of ‘realizations’ of the SSA, the number of reaction (and diffusion) events in stochastic solvers.
Syntax:
getNSteps()
Arguments: None
Return: int
- getTemp()
Return the simulation temperature.
Syntax:
getTemp()
Arguments: None
Return: float
- setTemp(t)
Set the simulation temperature. Currently, this will only influence the GHK flux rate, so will only influence simulations including membrane potential calculation.
Syntax:
setTemp(temp)
Arguments: float temp
Return: None
- setEfieldDT(efdt)
Set the stepsize for membrane potential solver (default 1us). This is the time for each voltage calculation step. The SSA will run until passing this stepsize, so in fact each membrane potential time step will vary slightly around the dt so as to be aligned with the SSA.
Syntax:
setEFieldDT(dt)
Arguments: float dt
Return: None
Compartment Data Access
- getCompVol(c)
Returns the volume of compartment with identifier string comp (in m^3).
Syntax:
getCompVol(comp)
Arguments: string comp
Return: float
- getCompCount(c, s)
DEPRECATED Returns the number of molecules of a species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined count from all tetrahedral elements in the compartment.
Syntax:
getCompCount(comp, spec)
Arguments: string comp string spec
Return: float
- setCompCount(c, s, n)
DEPRECATED Set the number of molecules of a species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined count from all tetrahedral elements in the compartment.
Syntax:
setCompCount(comp, spec, nspec)
Arguments: string comp string spec int nspec
Return: None
- getCompAmount(c, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined amount from all tetrahedral elements in the compartment.
Syntax:
getCompAmount(comp, spec)
Arguments: string comp string spec
Return: float
- setCompAmount(c, s, a)
DEPRECATED Set the amount (in mols) of species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined amount from all tetrahedral elements in the compartment.
Syntax:
setCompAmount(comp, spec, amount)
Arguments: string comp string spec float amount
Return: None
- getCompConc(c, s)
DEPRECATED Returns the concentration (in Molar units) of species with identifier string spec in compartment with identifier string comp.
Note: in a mesh-based simulation this is calculated from the combined number of molecules from all tetrahedral elements in the compartment and the total volume of the tetrahedrons.
Syntax:
getCompConc(comp, spec)
Arguments: string comp string spec
Return: float
- setCompConc(c, s, conc)
DEPRECATED Sets the concentration (in Molar units) of species with identifier string spec in compartment with identifier string comp to conc. In a discrete solver the continuous concentration is converted to a discrete number of molecules.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all tetrahedral elements in the compartment (i.e. a uniform distribution).
Syntax:
setCompConc(comp, spec, conc)
Arguments: string comp string spec float conc
Return: None
- getCompClamped(c, s)
DEPRECATED Returns True if species with identifier string spec in compartment with identifier string comp is clamped, which means the concentration remains the same regardless of reactions that consume or produce molecules of this species. Returns False if not.
Note: in a mesh-based simulation it returns True only if the species is clamped in all tetrahedral elements of the compartment.
Syntax:
getCompClamped(comp, spec)
Arguments: string comp string spec
Return: bool
- setCompClamped(c, s, b)
DEPRECATED Sets whether the concentration of species with identifier string spec in compartment with identifier string comp is clamped (clamped = True) or not (clamped = False). If a species is clamped the concentration stays the same regardless of reactions that consume or produce molecules of the species.
Note: in a mesh-based simulation this will set the species to be clamped or not in all tetrahedral elements of the compartment.
Syntax:
setCompClamped(comp, spec, clamped)
Arguments: string comp string spec bool clamped
Return: None
- getCompReacK(c, r)
Returns the macroscopic reaction constant of reaction with identifier string reac in compartment with identifier string comp. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation the value for the compartment is returned, although individual tetrahedral elements may have different values (set with setTetReacK).
Syntax:
getCompReacK(comp, reac)
Arguments: string comp string reac
Return: float
- setCompReacK(c, r, kf)
Sets the macroscopic reaction constant of reaction with identifier string reac in compartment with identifier string comp to kf. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation this method sets the reaction constant in all tetrahedral elements of the compartment to kf
Note: The default value still comes from the steps.model description, so calling reset() will return the reaction constant to that value.
Syntax:
setCompReacK(comp, reac, kf)
Arguments: string comp string reac float kf
Return: None
- getCompReacActive(c, r)
Returns whether a reaction with identifier string reac in compartment with identifier string comp is active (True) or not (False). If it’s not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this method will return True only if the reaction is active in all tetrahedral elements in the compartment.
Syntax:
getCompReacActive(comp, reac)
Arguments: string comp string reac
Return: bool
- setCompReacActive(c, r, a)
Activate (active = True) or deactivate (active = False) a reaction with identifier string reac in compartment with identifier string comp. If a reaction is not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this will activate/deactivate the reaction in all tetrahedral elements in the compartment.
Syntax:
setCompReacActive(comp, reac, active)
Arguments: string comp string reac bool active
Return: None
- getCompDiffD(c, d)
Returns the diffusion constant of diffusion rule with identifier string diff in compartment with identifier string comp. This constant is in units m^2/s.
Note: In a mesh-based solver the value for the compartment is returned, although individual or groups of tetrahedral elements may have different values (set with setTetDiffD).
Syntax:
getCompDiffD(comp, diff)
Arguments: string comp string diff
Return: float
- setCompDiffD(c, d, dcst)
Sets the diffusion constant of diffusion rule with identifier string diff in compartment with identifier string comp to dcst (in m^2/s).
Note: This method will set the diffusion constant in all tetrahedral elements in the compartment.
Note: The default value still comes from the steps.model description, so calling reset() will return the diffusion constants to that value.
Syntax:
setCompDiffD(comp, diff, dcst)
Arguments: string comp string diff float dcst
- Return:
None
- getCompDiffActive(c, d)
Returns whether a diffusion rule with identifier string diff in compartment with identifier string comp is active (True) or not (False). If diffusion of a species is inactive this means the molecules will remain in place and has the same effect as a diffusion constant of zero.
Syntax:
getCompDiffActive(comp, diff)
Arguments: string comp string diff
Return: bool
- setCompDiffActive(c, d, act)
Activate (active = True) or deactivate (active = False) a diffusion rule with identifier string diff in compartment with identifier string comp. If diffusion of a species is inactive this means the molecules will remain in place and is effectively the same as setting the diffusion constant to zero
Syntax:
setCompDiffActive(comp, diff, active)
Arguments: string comp string diff bool active
Return: None
- getCompReacC(c, r)
Returns the ‘stochastic reaction constant’ (or ‘specific probability rate constant’) of reaction with identifier string reac in compartment with identifier string comp.
The ‘stochastic reaction constant’ multiplied by infinitesimal time interval dt gives the average probability that one reaction channel of this reaction type will react accordingly in dt.
Note: in a mesh-based simulation (i.e. Tetexact), the stochastic reaction constant is computed as the weighted mean of the stochastic reaction constants in all tetrahedral elements of the compartment.
Syntax:
getCompReacC(comp, reac)
Arguments: string comp string reac
Return: float
- getCompReacH(c, r)
Returns h_mu, the distinct number of ways in which reaction with identifier string reac can occur in compartment with identifier string comp, by computing the product of its reactants. Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the h_mu’s over all tetrahedral elements in the compartment.
Syntax:
getCompReacH(comp, reac)
Arguments: string comp string reac
Return: float
- getCompReacA(c, r)
Returns the propensity of reaction with identifier string reac in compartment with identifier string comp.
The propensity of a reaction is a function of state and is defined as the function whose product with infinitesimal time dt gives the probability that the reaction will occur in the next dt. It is the ‘stochastic reaction constant’ multiplied by ‘h_mu’.
Note: in a mesh-based simulation (i.e. Tetexact), the propensity of a reaction in a compartment is computed as the sum of the propensities in all tetrahedral elements of the compartment.
Syntax:
getCompReacA(comp, reac)
Arguments: string comp string reac
Return: float
- getCompReacExtent(c, r)
Return the extent of reaction with identifier string reac in compartment with identifier string comp, that is the number of times the reaction has occurred up to the current simulation time.
Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the reaction extents in all tetrahedral elements of the compartment.
Syntax:
getCompReacExtent(comp, reac)
Arguments: string comp string reac
Return: index_t
- resetCompReacExtent(c, r)
Resets the extent of reaction with identifier string reac in compartment with identifier string comp to zero.
Note: in a mesh-based simulation (i.e. Tetexact), resets the extents of the reaction in all tetrahedral elements of the compartment.
Syntax:
resetCompReacExtent(comp, reac)
Arguments: string comp string reac
Return: None
Patch Data Access
- getPatchArea(p)
Returns the area of patch with identifier string patch (in m^2).
Syntax:
getPatchArea(patch)
Arguments: string patch
Return: float
- getPatchCount(p, s)
DEPRECATED Returns the number of molecules of species with identifier string spec in patch with identifier string patch.Note: in a mesh-based simulation this is the combined count from all triangular elements in the patch.
Syntax:
getPatchCount(patch, spec)
Arguments: string patch string spec
Return: float
- setPatchCount(p, s, n)
DEPRECATED Sets the number of molecules of species with identifier string spec in patch with identifier string patch to n. Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
setPatchCount(patch, spec, n)
Arguments: string patch string spec int n
Return: float
- getPatchAmount(p, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in patch with identifier string patch.
Note: in a mesh-based simulation this is the combined amount from all triangular elements in the patch.
Syntax:
getPatchAmount(patch, spec)
Arguments: string patch string spec
Return: float
- setPatchAmount(p, s, a)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in patch with identifier string patch to a. In a discrete solver, such as Wmdirect and Tetexact, this continuous value is converted internally into a discrete number of molecules by multiplication with Avogadro’s number.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
setPatchAmount(patch, spec, a)
Arguments: string patch string spec float a
Return: None
- getPatchClamped(p, s)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in patch with identifier string patch to a. In a discrete solver, such as Wmdirect and Tetexact, this continuous value is converted internally into a discrete number of molecules by multiplication with Avogadro’s number.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
getPatchClamped(patch, spec)
Arguments: string patch string spec
Return: bool
- setPatchClamped(p, s, buf)
DEPRECATED Sets whether the species with identifier string spec in patch with identifier string patch is clamped (clamped = True) or not (clamped = False). If a species is clamped the number of molecules stays the same regardless of surface reactions that consume or produce molecules of the species.
Note: in a mesh-based simulation this will set the species to be clamped in all triangular elements of the patch.
Syntax:
setPatchClamped(patch, spec, clamped)
Arguments: string patch string spec bool clamped
Return: None
- getPatchSReacK(p, r)
Returns the macroscopic reaction constant of surface reaction with identifier string sreac in patch with identifier string patch. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based solver the value for the patch is returned, although individual triangle elements may have different values (set with setTriSReacK).
Syntax:
getPatchSReacK(patch, reac)
Arguments: string patch string reac
Return: float
- setPatchSReacK(p, r, kf)
Sets the macroscopic reaction constant of surface reaction with identifier string sreac in patch with identifier string patch to kf. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation this method sets the surface reaction constant in all triangular elements of the patch to kf.
Note: The default value still comes from the steps.model description, so calling reset() will return the surface reaction constant to that value.
Syntax:
setPatchSReacK(patch, reac, kf)
Arguments: string patch string reac float kf
Return: None
- getPatchSReacActive(p, r)
Returns whether a surface reaction with identifier string sreac in patch with identifier string patch is active (True) or not (False). If it’s not active this means that a surface reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this method will return True only if the surface reaction is active in all triangular elements in the patch.
Syntax:
getPatchSReacActive(patch, reac)
Arguments: string patch string reac
Return: bool
- setPatchSReacActive(p, r, a)
Activate (active = True) or deactivate (active = False) a surface reaction with identifier string sreac in patch with identifier string patch. If a surface reaction is not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this will activate/ deactivate the reaction in all triangular elements in the patch.
Syntax:
setPatchSReacActive(patch, reac, active)
Arguments: string patch string reac bool active
Return: None
- getPatchSReacC(p, r)
Returns the ‘stochastic reaction constant’ (or ‘specific probability rate constant’) of surface reaction with identifier string sreac in patch with identifier string patch.
Note: in a mesh-based simulation (i.e. Tetexact), the stochastic reaction constant is computed as the weighted mean of the stochastic reaction constants in all triangular elements of the patch.
Syntax:
getPatchSReacC(patch, reac)
Arguments: string patch string reac
Return: float
- getPatchSReacH(p, r)
Returns h_mu, the distinct number of ways in which surface reaction with identifier string sreac can occur in patch with identifier string patch, by computing the product of its reactants. Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the h_mu’s over all triangular elements in the patch.
Syntax:
getPatchSReacH(patch, reac)
Arguments: string patch string reac
Return: float
- getPatchSReacA(p, r)
Returns the propensity of surface reaction with identifier string sreac in patch with identifier string patch. Note: in a mesh-based simulation (i.e. Tetexact), the propensity of a surface reaction in a patch is computed as the sum of the propensities in all triangular elements of the patch.
Syntax:
getPatchSReacA(patch, reac)
Arguments: string patch string reac
Return: float
- getPatchSReacExtent(p, r)
Returns the extent of surface reaction with identifier string sreac in patch with identifier string patch, that is the number of times the surface reaction has occurred up to the current simulation time.
Note: in a mesh-based simulation (i.e. Tetexact), returns the sum of the reaction extents in all triangular elements of the patch.
Syntax:
getPatchSReacExtent(patch, reac)
Arguments: string patch string reac
Return: index_t
- resetPatchSReacExtent(p, r)
Resets the extent of reaction with identifier string sreac in patch with identifier string patch to zero.
Note: in a mesh-based simulation (i.e. Tetexact), resets the extents of the reaction in all triangular elements of the patch.
Syntax:
resetPatchSReacExtent(patch, reac)
Arguments: string patch string reac
Return: None
- getPatchVDepSReacActive(p, vsr)
Returns whether a voltage-dependent surface reaction with identifier string vsreac in patch with identifier string patch is active (True) or not (False). If it’s not active this means that the voltage-dependent surface reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this method will return True only if the voltage-dependent surface reaction is active in all triangular elements in the patch.
Syntax:
getPatchVDepSReacActive(patch, vsreac)
Arguments: string patch string vsreac
Return: bool
- setPatchVDepSReacActive(p, vsr, a)
Activate (active = True) or deactivate (active = False) a voltage-dependent surface reaction with identifier string vsreac in patch with identifier string patch. If a voltage-dependent surface reaction is not active this means that a reaction will never occur regardless of whether the reactants are present in sufficient numbers or not.
Note: In a mesh-based simulation this will activate/ deactivate the reaction in all triangular elements in the patch.
Syntax:
setPatchVDepSReacActive(patch, vsreac, active)
Arguments: string patch string vsreac bool active
Return: None
Diffusion Boundary Data Access
- setDiffBoundaryDiffusionActive(db, s, act)
DEPRECATED Activates or inactivates diffusion across a diffusion boundary for a species.
Syntax:
setDiffBoundaryDiffusionActive(diffb, spec, act)
Arguments: string diffb string spec bool act
Return: None
- getDiffBoundaryDiffusionActive(db, s)
DEPRECATED Returns whether diffusion is active across a diffusion boundary for a species.
Syntax:
getDiffBoundaryDiffusionActive(diffb, spec)
Arguments: string diffb string spec
Return: bool
- setDiffBoundaryDcst(db, s, dcst, direction_comp='')
DEPRECATED Set the diffusion constant of tetrahedrons across a diffusion boundary. If direction_comp is provided, only set dcsts of diffusion towards it (Directional dcsts of diffusions in tetrahedrons in the other compartment of the diffusion boundary towards tetrahedons in the direction compartment).
Syntax:
setDiffBoundaryDcst(diffb, spec, dcst, direction_comp = '')
Arguments: string diffb string spec float dcst string direction_comp
Return: None
Surface Diffusion Boundary Data Access
- setSDiffBoundaryDiffusionActive(sdb, s, act)
DEPRECATED Activates or inactivates diffusion across a surface diffusion boundary for a species.
Syntax:
setSDiffBoundaryDiffusionActive(sdiffb, spec, act)
Arguments: string sdiffb string spec bool act
Return: None
- getSDiffBoundaryDiffusionActive(sdb, s)
DEPRECATED Returns whether diffusion is active across a surface diffusion boundary for a species.
Syntax:
getSDiffBoundaryDiffusionActive(sdiffb, spec)
Arguments: string sdiffb string spec
Return: bool
- setSDiffBoundaryDcst(sdb, s, dcst, direction_patch='')
DEPRECATED Set the diffusion constant of triangles across a surface diffusion boundary. If direction_patch is provided, only set dcsts of diffusion towards it (Directional dcsts of diffusions in triangles in the other patches of the diffusion boundary towards triangles in the direction patch).
Syntax:
setSDiffBoundaryDcst(sdiffb, spec, dcst, direction_patch = '')
Arguments: string sdiffb string spec float dcst string direction_patch
Return: None
Tetrahedral Data Access
- getTetVol(idx)
Returns the volume (in m^3) of the tetrahedral element with index idx.
Syntax:
getTetVol(idx)
Arguments: index_t idx
Return: float
- getTetSpecDefined(idx, s)
Returns whether species with identifier string spec is defined in the tetrahedral element with index idx.
Syntax:
getTetSpecDefined(idx, spec)
Arguments: index_t idx string spec
Return: bool
- getTetCount(idx, s)
DEPRECATED Returns the number of molecules of species with identifier string spec in the tetrahedral element with index idx.
Syntax:
getTetCount(idx, spec)
Arguments: index_t idx string spec
Return: int
- setTetCount(idx, s, n)
DEPRECATED Sets the number of molecules of species with identifier string spec in tetrahedral element with index idx to n.
Syntax:
setTetCount(idx, spec, n)
Arguments: index_t idx string spec int n
Return: None
- getTetAmount(idx, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in tetrahedral element with index idx.
Syntax:
getTetAmount(idx, spec)
Arguments: index_t idx string spec
Return: float
- setTetAmount(idx, s, m)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in tetrahedral element with index idx to a. This continuous value must be converted internally to a discrete number of molecules by multiplication with Avogadro’s number.
Due to the small volumes of tetrahedral elements the difference between ‘rounding up’ and ‘rounding down’ can be a significant difference in concentration.
Syntax:
setTetAmount(idx, spec, a)
Arguments: index_t idx string spec float a
Return: None
- getTetConc(idx, s)
DEPRECATED Returns the concentration (in Molar units) of species with identifier string spec in a tetrahedral element with index idx.
Syntax:
getTetConc(idx, spec)
Arguments: index_t idx string spec
Return: float
- setTetConc(idx, s, c)
DEPRECATED Sets the concentration (in Molar units) of species with identifier string spec in a tetrahedral element with index idx to conc.This continuous value must be converted internally to a discrete number of molecules.
Due to the small volumes of tetrahedral elements the difference between ‘rounding up’ and ‘rounding down’ can be a large difference in concentration.
Syntax:
setTetConc(idx, spec, conc)
Arguments: index_t idx string spec float conc
Return: None
- getTetClamped(idx, s)
DEPRECATED Returns True if concentration of species with identifier string spec in tetrahedral element with index idx is clamped, which means the concentration stays the same regardless of reactions that consume or produce molecules of this species or diffusion of this species into or out of the tetrahedral element. Returns False if not.
Syntax:
getTetClamped(idx, spec)
Arguments: index_t idx string spec
Return: bool
- setTetClamped(idx, s, buf)
DEPRECATED Sets whether the concentration of species spec in tetrahedral element with index idx is clamped (clamped = True) or not (clamped = False). If a species is clamped the concentration stays the same regardless of reactions that consume or produce molecules of the species or diffusion of the species into or out of the tetrahedral element.
Syntax:
setTetClamped(idx, spec, clamped)
Arguments: index_t idx string spec bool clamped
Return: None
- getTetReacK(idx, r)
Returns the macroscopic reaction constant of reaction with identifier string reac in tetrahedral element with index idx. The unit of the reaction constant depends on the order of the reaction.
Syntax:
getTetReacK(idx, reac)
Arguments: index_t idx string reac
Return: float
- setTetReacK(idx, r, kf)
Sets the macroscopic reaction constant of reaction with identifier string reac in tetrahedral element with index idx to kf. The units of the reaction constant depends on the order of the reaction.
Syntax:
setTetReacK(idx, reac, kf)
Arguments: index_t idx string reac float kf
Return: None
- getTetReacActive(idx, r)
Returns whether reaction with identifier string reac in tetrahedral element with index idx is active (True) or not (False). If it’s not active this means that the reaction will never occur regardless of whether reactants are present in sufficient numbers or not.
Syntax:
getTetReacActive(idx, reac)
Arguments: index_t idx string reac
Return: bool
- setTetReacActive(idx, r, act)
Activate (active = True) or deactivate (active = False) a reaction with identifier string reac in tetrahedral element with index idx. If it’s not active this means that the reaction will never occur regardless of whether reactants are present in sufficient numbers or not.
Syntax:
setTetReacActive(idx, reac, active)
Arguments: index_t idx string reac bool active
Return: None
- getTetDiffD(idx, d, direction_tet=UNKNOWN_TET)
Returns the diffusion constant of diffusion rule with identifier string diff in tetrahedral element with index idx. This constant is in units m^2/s. If direction_tet is specified, return the diffusion constant towards that direction.
Syntax:
getTetDiffD(idx, diff, direction_tet = UNKNOWN_TET)
Arguments: index_t idx string diff direction_tet
Return: float
- setTetDiffD(idx, d, dk, direction_tet=UNKNOWN_TET)
Sets the diffusion constant of diffusion rule with identifier string diff in tetrahedral element with index idx to dcst (in m^2/s). Specify direction_tet to set the constant only towards a given tetrahedron direction. Syntax:
setTetDiffD(idx, diff, dcst, direction_tet = UNKNOWN_TET)
Arguments: index_t idx string diff float dcst index_t direction_tet
Return: None
- getTetDiffActive(idx, d)
Returns whether diffusion with identifier string diff in tetrahedral element with index idx is active (True) or not (False). If diffusion of a species is inactive this means the molecules will never diffuse out of the tetrahedron and has the same effect as a diffusion constant of zero.
Syntax:
getTetDiffActive(idx, diff)
Arguments: index_t idx string diff
Return: bool
- setTetDiffActive(idx, d, act)
Activate (active = True) or deactivate (active = False) diffusion rule with identifier string diff in tetrahedral element with index idx. If diffusion of a species is inactive this means the molecules will never diffuse out of the tetrahedron and has the same effect as a diffusion constant of zero.
Syntax:
setTetDiffActive(idx, diff, active)
Arguments: index_t idx string diff bool active
Return: None
- getTetReacC(idx, r)
Returns the ‘stochastic reaction constant’ (or ‘specific probability rate constant’) of reaction with identifier string reac in tetrahedral element with index idx.
Syntax:
getTetReacC(idx, reac)
Arguments: index_t idx string reac
Return: float
- getTetReacH(idx, r)
Returns h_mu, the distinct number of ways in which reaction with identifier string reac can occur in tetrahedral element with index idx, by computing the product of its reactants.
Syntax:
getTetReacH(idx, reac)
Arguments: index_t idx string reac
Return: float
- getTetReacA(idx, r)
Returns the propensity of reaction with identifier string reac in tetrahedral element with index idx.
Syntax:
getTetReacA(idx, reac)
Arguments: index_t idx string reac
Return: float
- getTetDiffA(idx, d)
Returns the propensity of diffusion rule with identifier string diff in tetrahedral element with index idx.
Syntax:
getTetDiffA(idx, reac)
Arguments: index_t idx string reac
Return: float
- getTetV(idx)
Returns the potential (in volts) of tetrahedral element with index idx, taken at the barycenter.
Syntax:
getTetV(idx)
Arguments: index_t idx
Return: float
- setTetV(idx, v)
Set the potential (in volts) of tetrahedral element with index idx.
Syntax:
setTetV(idx, v)
Arguments: index_t idx float v
Return: None
- getTetVClamped(idx)
Returns true if the potential of tetrahedral element with index idx is clamped to some voltage.
Syntax:
getTetVClamped(idx)
Arguments: index_t idx
Return: bool
- setTetVClamped(idx, cl)
Sets whether the potential of tetrahedral element with index idx is clamped (clamped = True) or not (clamped = False).
Syntax:
setTetVClamped(idx, clamped)
Arguments: index_t idx bool clamped
Return: None
Triangular Data Access
- getTriArea(idx)
Returns the area (in m^2) of the triangular element with index idx.
Syntax:
getTriArea(idx)
Arguments: index_t idx
Return: float
- getTriSpecDefined(idx, s)
Returns whether species with identifier string spec is defined in the triangle element with index idx.
Syntax:
getTriSpecDefined(idx, spec)
Arguments: index_t idx string spec
Return: bool
- getTriCount(idx, s)
DEPRECATED Returns the number of molecules of species with identifier string spec in the triangular element with index idx.
Syntax:
getTriCount(idx, spec)
Arguments: index_t idx string spec
Return: float
- setTriCount(idx, s, n)
DEPRECATED Sets the number of molecules of species with identifier string spec in triangular element with index idx to n.
Syntax:
setTriCount(idx, spec, n)
Arguments: index_t idx string spec int n
Return: None
- getTriAmount(idx, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in triangular element with index idx.
Syntax:
getTriAmount(idx, spec)
Arguments: index_t idx string spec
Return: float
- setTriAmount(idx, s, m)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in triangular element with index idx to a. This continuous value must be converted internally to a discrete number of molecules by multiplication with Avogadro’s number.
Syntax:
setTriAmount(idx, spec, a)
Arguments: index_t idx string spec float a
Return: None
- getTriClamped(idx, s)
DEPRECATED Returns True if the species with identifier string spec in triangular element with index idx is clamped, which means the number of molecules stays the same regardless of reactions that consume or produce molecules of this species. Returns False if not.
Syntax:
getTriClamped(idx, spec)
Arguments: index_t idx string spec
Return: bool
- setTriClamped(idx, s, buf)
DEPRECATED Sets whether the concentration of species spec in triangular element with index idx is clamped (clamped = True) or not (clamped = False). If a species is clamped the concentration stays the same regardless of reactions that consume or produce molecules of the species.
Syntax:
setTriClamped(idx, spec, clamped)
Arguments: index_t idx string spec bool clamped
Return: None
- getTriSReacK(idx, r)
Returns the macroscopic reaction constant of surface reaction with identifier string sreac in triangular element with index idx. The units of the reaction constant depends on the order of the reaction.
Syntax:
getTriSReacK(idx, reac)
Arguments: index_t idx string reac
Return: float
- setTriSReacK(idx, r, kf)
Sets the macroscopic reaction constant of surface reaction with identifier string sreac in triangular element with index idx to kf. The units of the reaction constant depends on the order of the reaction.
Syntax:
setTriSReacK(idx, reac, kf)
Arguments: index_t idx string reac float kf
Return: None
- getTriSReacActive(idx, r)
Returns whether surface reaction with identifier string sreac in triangular element with index idx is active (True) or not (False). If it’s not active this means that the surface reaction will never occur regardless of whether reactants are present in sufficient numbers or not.
Syntax:
getTriSReacActive(idx, reac)
Arguments: index_t idx string reac
Return: bool
- setTriSReacActive(idx, r, act)
Activate (active = True) or deactivate (active = False) a surface reaction with identifier string sreac in triangular element with index idx. If it’s not active this means that the surface reaction will never occur regardless of whether reactants are present in sufficient numbers or not.
Syntax:
setTriSReacActive(idx, reac, active)
Arguments: index_t idx string reac bool active
Return: None
- getTriSReacC(idx, r)
Returns the ‘stochastic reaction constant’ (or ‘specific probability rate constant’) of surface reaction with identifier string sreac in triangular element with index idx.
Syntax:
getTriSReacC(idx, reac)
Arguments: index_t idx string reac
Return: float
- getTriSReacH(idx, r)
Returns h_mu, the distinct number of ways in which surface reaction with identifier string sreac can occur in triangular element with index idx, by computing the product of its reactants.
Syntax:
getTriSReacH(idx, reac)
Arguments: index_t idx string reac
Return: float
- getTriSReacA(idx, r)
Returns the propensity of surface reaction with identifier string sreac in triangular element with index idx.
Syntax:
getTriSReacA(idx, reac)
Arguments: index_t idx string reac
Return: float
- getTriSDiffD(idx, d, direction_tri=UNKNOWN_TRI)
Returns the diffusion constant of diffusion rule with identifier string diff in triangle element with index idx. If direction_tri is specified, return the diffusion constant towards that direction.
Syntax:
getTriDiffD(idx, diff, direction_tri = UNKNOWN_TRI)
Arguments: index_t idx string diff index_t direction_tri
Return: float
- setTriSDiffD(idx, d, dk, direction_tri=UNKNOWN_TRI)
Sets the diffusion constant of diffusion rule with identifier string diff in triangle element with index idx to dcst. Specify direction_tri to set the constant only towards a given triangle direction. Syntax:
setTriSDiffD(idx, diff, dcst, direction_tri = UNKNOWN_TRI)
Arguments: index_t idx string diff float dcst index_t direction_tri
Return: None
- getTriV(idx)
Returns the potential (in volts) of triangle element with index idx, taken at the barycenter.
Syntax:
getTriV(idx)
Arguments: index_t idx
Return: float
- setTriV(idx, v)
Set the potential (in volts) of triangle element with index idx.
Syntax:
setTriV(idx, v)
Arguments: index_t idx float v
Return: None
- getTriVClamped(idx)
Returns true if the potential of triangle element with index idx is clamped to some voltage.
Syntax:
getTriVClamped(idx)
Arguments: index_t idx
Return: bool
- setTriVClamped(idx, cl)
Sets whether the potential of triangle element with index idx is clamped (clamped = True) or not (clamped = False).
Syntax:
setTriVClamped(idx, clamped)
Arguments: index_t idx bool clamped
Return: None
- getTriOhmicI(idx, oc='')
Returns the ohmic current of triangle element with index idx, in amps.
Syntax:
getTriOhmicI(idx, oc)
Arguments: index_t idx string oc (default = ‘’)
Return: float
- getTriGHKI(idx, ghk='')
Returns the GHK current of triangle element with index idx, in amps.
Syntax:
getTriGHKI(idx, ghk)
Arguments: index_t idx string ghk (default = ‘’)
Return: float
- getTriI(idx)
Returns the current of triangle element with index idx, in amps, at the last EField calculation step.
Syntax:
getTriI(idx)
Arguments: index_t idx
Return: float
- setTriIClamp(idx, i)
Set current clamp to triangle element with index idx to current i (amps). NOTE: Convention is maintained that a positive current clamp is depolarizing, a negative current clamp is hyperpolarizing.
Syntax:
setTriIClamp(idx, i)
Arguments: index_t idx float i
Return: None
- getTriVDepSReacActive(idx, vsr)
Returns whether voltage-dependent surface reaction with identifier string vsreac in triangular element with index idx is active (True) or not (False). If it’s not active this means that the voltage-dependent surface reaction will never occur regardless of whether reactants are present in sufficient numbers or not.
Syntax:
getTriVDepSReacActive(idx, reac)
Arguments: index_t idx string vsreac
Return: bool
- setTriVDepSReacActive(idx, vsr, act)
Activate (active = True) or deactivate (active = False) a voltage-dependent surface reaction with identifier string vsreac in triangular element with index idx. If it’s not active this means that the voltage-dependent surface reaction will never occur regardless of whether reactants are present in sufficient numbers or not.
Syntax:
setTriVDepSReacActive(idx, vsreac, active)
Arguments: index_t idx string vsreac bool active
Return: None
Vertex Data Access
- getVertV(vidx)
Returns the potential (in volts) of vertex element with index idx.
Syntax:
getVertV(idx)
Arguments: index_t idx
Return: float
- setVertV(vidx, v)
Set the potential (in volts) of vertex element with index idx.
Syntax:
setVertV(idx, v)
Arguments: index_t idx float v
Return: None
- getVertVClamped(vidx)
Returns true if the potential of vertex element with index idx is clamped to some voltage.
Syntax:
getVertVClamped(idx)
Arguments: index_t idx
Return: bool
- setVertVClamped(vidx, cl)
Sets whether the potential of vertex element with index idx is clamped (clamped = True) or not (clamped = False).
Syntax:
setVertVClamped(idx, clamped)
Arguments: index_t idx bool clamped
Return: None
- setVertIClamp(vidx, i)
Set current clamp to vertex element with index idx to current i (Amps). NOTE: Convention is maintained that a positive current clamp is depolarizing, a negative current clamp is hyperpolarizing.
Syntax:
setVertIClamp(idx, i)
Arguments: index_t idx float i
Return: None
Membrane Data Access
- saveMembOpt(opt_file_name)
Saves the vertex optimization in the Efield structure.
Syntax:
saveMembOpt()
Arguments: string filename
Return: None
- setMembPotential(m, v)
Sets the potential (in volts) of membrane with string identifier memb. NOTE: This method will set the potential of all nodes in the volume conductor to the same value.
Syntax:
setMembPotential(memb, v)
Arguments: string memb float v
Return: None
- setMembCapac(m, cm)
Sets the specific membrane capacitance (in farad / m^2) of membrane with string identifier memb.
Syntax:
setMembCapac(memb, cm)
Arguments: string memb float cm
Return: None
- setMembVolRes(m, ro)
Sets the bulk electrical resistivity (in ohm.m) of the volume conductor assocaited with membrane with string identifier memb.
Syntax:
setMembVolRes(memb, ro)
Arguments: string memb float ro
Return: None
- setMembRes(m, ro, vrev)
Sets the surface electrical resistivity ro (in ohm.m^2) of the membrane with string identifier memb. Reversal potential vrev is required in Volts.
Syntax:
setMembRes(memb, ro, vrev)
Arguments: string memb float ro float vrev
Return: None
Batch Data Access
- getBatchTetCounts(tets, s)
DEPRECATED Get the counts of a species s in a list of tetrahedrons.
Syntax:
getBatchTetCounts(tets, s)
Arguments: list<index_t> tets string s
Return: list<double>
- getBatchTriCounts(tris, s)
DEPRECATED Get the counts of a species s in a list of triangles.
Syntax:
getBatchTriCounts(tris, s)
Arguments: list<index_t> tris string s
Return: list<double>
- getBatchTetCountsNP(indices, s, counts)
DEPRECATED Get the counts of a species s in a list of tetrahedrons.
- Syntax::
getBatchTetCountsNP(indices, s, counts)
Arguments: numpy.array<index_t> indices string s numpy.array<double, length = len(indices)>
Return: None
- getBatchTriCountsNP(indices, s, counts)
DEPRECATED Get the counts of a species s in a list of triangles.
- Syntax::
getBatchTriCountsNP(indices, s, counts)
Arguments: numpy.array<index_t> indices string s numpy.array<double, length = len(indices)>
Return: None
Region of Interest functions
- getROITetCounts(roi, s)
DEPRECATED Get the counts of a species s in tetrehedrons of a ROI.
Syntax:
getROITetCounts(roi, s)
Arguments: string roi string s
Return: list<float>
- getROITriCounts(roi, s)
DEPRECATED Get the counts of a species s in triangles of a ROI.
Syntax:
getROITriCounts(roi, s)
Arguments: string roi string s
Return: list<float>
- getROITetCountsNP(roi, s, counts)
DEPRECATED Get the counts of a species s in tetrehedrons of a ROI.
- Syntax::
getROITetCountsNP(roi, s, counts)
Arguments: string roi string s numpy.array<float, length = len(indices)>
Return: None
- getROITriCountsNP(roi, s, counts)
DEPRECATED Get the counts of a species s in triangles of a ROI.
- Syntax::
getROITriCountsNP(roi, s, counts)
Arguments: string roi string s numpy.array<float, length = len(indices)>
Return: None
- getROIVol(roi)
Get the volume of a ROI.
- Syntax::
getROIVol(roi)
Arguments: string roi
Return: float
- getROIArea(roi)
Get the area of a ROI.
- Syntax::
getROIArea(roi)
Arguments: string roi
Return: float
- getROICount(roi, s)
DEPRECATED Get the count of a species in a ROI.
- Syntax::
getROICount(roi, s)
Arguments: string roi string s
Return: float
- setROICount(roi, s, count)
DEPRECATED Set the count of a species in a ROI.
- Syntax::
setROICount(roi, s, count)
Arguments: string roi string s float count
Return: None
- getROIAmount(roi, s)
DEPRECATED Get the amount of a species in a ROI.
- Syntax::
getROIAmount(roi, s)
Arguments: string roi string s
Return: float
- getROIConc(roi, s)
DEPRECATED Get the concentration of a species in a ROI.
- Syntax::
getROIConc(roi, s, count)
Arguments: string roi string s
Return: float
- setROIClamped(roi, s, b)
DEPRECATED Set a species in a ROI to be clamped or not. The count of species s in the ROI is clamped if b is True, not clamped if b is False.
- Syntax::
setROIClamped(roi, s, b)
Arguments: string roi string s bool b
Return: None
- setROIReacK(roi, r, kf)
Sets the macroscopic reaction constant of reaction with identifier string r in a ROI with identifier string roi to kf. The unit of the reaction constant depends on the order of the reaction.
Note: The default value still comes from the steps.model description, so calling reset() will return the reaction constant to that value.
- Syntax::
setROIReacK(roi, r, kf)
Arguments: string roi string r float kf
Return: None
- setROISReacK(roi, sr, kf)
Sets the macroscopic reaction constant of surface reaction with identifier string sr in a ROI with identifier string roi to kf. The unit of the reaction constant depends on the order of the reaction.
Note: The default value still comes from the steps.model description, so calling reset() will return the reaction constant to that value.
- Syntax::
setROISReacK(roi, sr, kf)
Arguments: string roi string sr float kf
Return: None
- setROIDiffD(roi, diff, dcst)
Sets the macroscopic diffusion constant of diffusion with identifier string diff in a ROI with identifier string roi to dcst.
Note: The default value still comes from the steps.model description, so calling reset() will return the diffusion constant to that value.
- Syntax::
setROIDiffD(roi, diff, dcst)
Arguments: string roi string diff float dcst
- Return:
None
- setROIReacActive(roi, r, a)
Set reaction r in a ROI to be active or not.
- Syntax::
setROIReacActive(roi, r, a)
Arguments: string roi string r bool a
Return: None
- setROISReacActive(roi, sr, a)
Set surface reaction sr in a ROI to be active or not.
- Syntax::
setROISReacActive(roi, sr, a)
Arguments: string roi string sr bool a
Return: None
- setROIDiffActive(roi, d, act)
Set diffusion d in a ROI to be active or not.
- Syntax::
setROIDiffActive(roi, sr, a)
Arguments: string roi string sr bool a
Return: None
- setROIVDepSReacActive(roi, vsr, a)
Set voltage dependent surface reaction vsr in a ROI to be active or not.
- Syntax::
setROIVDepSReacActive(roi, vsr, a)
Arguments: string roi string vsr bool a
Return: None
- getROIReacExtent(roi, r)
Return the extent of reaction with identifier string reac in ROI with identifier string roi, that is the number of times the reaction has occurred up to the current simulation time.
- Syntax::
getROIReacExtent(roi, reac)
Arguments: string roi string r
Return: index_t
- resetROIReacExtent(roi, r)
Reset the extent of reaction with identifier string reac in ROI with identifier string roi, that is the number of times the reaction has occurred up to the current simulation time, to 0.
- Syntax::
resetROIReacExtent(roi, reac)
Arguments: string roi string r
Return: None
- getROISReacExtent(roi, sr)
Return the extent of surface reaction with identifier string sreac in ROI with identifier string roi, that is the number of times the reaction has occurred up to the current simulation time.
- Syntax::
getROISReacExtent(roi, sreac)
Arguments: string roi string sr
Return: index_t
- resetROISReacExtent(roi, sr)
Reset the extent of surface reaction with identifier string reac in ROI with identifier string roi, that is the number of times the reaction has occurred up to the current simulation time, to 0.
- Syntax::
resetROISReacExtent(roi, reac)
Arguments: string roi string sr
Return: None
- getROIDiffExtent(roi, d)
Return the extent of diffusion with identifier string diff in ROI with identifier string roi, that is the number of times the diffusion has occurred up to the current simulation time.
- Syntax::
getROIDiffExtent(roi, diff)
Arguments: string roi string d
Return: index_t
- resetROIDiffExtent(roi, s)
Reset the extent of diffusion with identifier string diff in ROI with identifier string roi, that is the number of times the diffusion has occurred up to the current simulation time, to 0.
- Syntax::
resetROIDiffExtent(roi, diff)
Arguments: string roi string d
Return: None
- class TetODE[source]
Construction:
sim = steps.solver.TetODE(model, geom, rng=None, calcMembPot = 0)
Create a spatial determinstic solver based on the CVODE library. If voltage is to be simulated, argument calcMemPot=1 will set to the default solver. calcMembPot=0 means voltage will not be simulated.
Arguments: steps.model.Model model steps.geom.Geom geom steps.rng.RNG rng (default=None) int calcMemPot (default=0)
Construction:
sim = steps.solver.TetODE(model, geom, rng=None, calcMembPot = 0, calcMembPot_lenient = False)
Create a spatial determinstic solver based on the CVODE library. If voltage is to be simulated, argument calcMemPot=1 will set to the default solver. calcMembPot=0 means voltage will not be simulated.
Arguments: steps.model.Model model steps.geom.Geom geom steps.rng.RNG rng (default=None) int calcMembPot (default=0) bool calcMembPot_lenient (default=False)
Solver Information
- getSolverName()
Returns a string of the solver’s name.
Syntax:
getSolverName()
Arguments: None
Return: string
- getSolverDesc()
Returns a string giving a short description of the solver.
Syntax:
getSolverDesc()
Arguments: None
Return: string
- getSolverAuthors()
Returns a string of the solver authors names.
Syntax:
getSolverAuthors()
Arguments: None
Return: string
- getSolverEmail()
Returns a string giving the author’s email address.
Syntax:
getSolverEmail()
Arguments: None
Return: string
Solver Control
- checkpoint(file_name)
Checkpoint data to a file.
Syntax:
checkpoint(file_name)
Arguments: string file_name
Return: None
- restore(file_name)
Restore data from a file.
Syntax:
restore(file_name)
Arguments: string file_name
Return: None
- reset()
Reset the simulation to the state the solver was initialised to. Typically, this resets all concentrations of all chemical species in all elements (whether compartments and patches in a well-mixed solver or tetrahedrons and triangles in a mesh-based solver) to zero, resets the simulation time to zero and resets reaction (and diffusion) rates to the default values described in the steps.model objects. All reaction (and diffusion) rules are reset to active and all compartment volumes and patch areas are reset to default values described in steps.geom objects (for well-mixed solvers). Usually, this method should be called before starting each simulation iteration.
Syntax:
reset()
Arguments: None
Return: None
- getTime()
Returns the current simulation time in seconds.
Syntax:
getTime()
Arguments: None
Return: float
- run(end_time, cp_interval=0.0, prefix='')[source]
Run the simulation until <end_time>, automatically checkpoint at each <cp_interval>. Prefix can be added using prefix=<prefix_string>.
- advance(advance_time, cp_interval=0.0, prefix='')[source]
Advance the simulation for <advance_time>, automatically checkpoint at each <cp_interval>. Prefix can be added using prefix=<prefix_string>.
- getTemp()
Return the simulation temperature.
Syntax:
getTemp()
Arguments: None
Return: float
- setTemp(t)
Set the simulation temperature. Currently, this will only influence the GHK flux rate, so will only influence simulations including membrane potential calculation.
Syntax:
setTemp(temp)
Arguments: float temp
Return: None
Compartment Data Access
- getCompCount(c, s)
DEPRECATED Returns the number of molecules of a species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined count from all tetrahedral elements in the compartment.
Syntax:
getCompCount(comp, spec)
Arguments: string comp string spec
Return: float
- setCompCount(c, s, n)
DEPRECATED Set the number of molecules of a species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined count from all tetrahedral elements in the compartment.
Syntax:
setCompCount(comp, spec, nspec)
Arguments: string comp string spec int nspec
Return: None
- getCompVol(c)
Returns the volume of compartment with identifier string comp (in m^3).
Syntax:
getCompVol(comp)
Arguments: string comp
Return: float
- getCompAmount(c, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined amount from all tetrahedral elements in the compartment.
Syntax:
getCompAmount(comp, spec)
Arguments: string comp string spec
Return: float
- setCompAmount(c, s, a)
DEPRECATED Set the amount (in mols) of species with identifier string spec in compartment with identifier string comp.
In a mesh-based simulation this is the combined amount from all tetrahedral elements in the compartment.
Syntax:
setCompAmount(comp, spec, amount)
Arguments: string comp string spec float amount
Return: None
- getCompConc(c, s)
DEPRECATED Returns the concentration (in Molar units) of species with identifier string spec in compartment with identifier string comp.
Note: in a mesh-based simulation this is calculated from the combined number of molecules from all tetrahedral elements in the compartment and the total volume of the tetrahedrons.
Syntax:
getCompConc(comp, spec)
Arguments: string comp string spec
Return: float
- setCompConc(c, s, conc)
DEPRECATED Sets the concentration (in Molar units) of species with identifier string spec in compartment with identifier string comp to conc. In a discrete solver the continuous concentration is converted to a discrete number of molecules.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all tetrahedral elements in the compartment (i.e. a uniform distribution).
Syntax:
setCompConc(comp, spec, conc)
Arguments: string comp string spec float conc
Return: None
- setCompReacK(c, r, kf)
Sets the macroscopic reaction constant of reaction with identifier string reac in compartment with identifier string comp to kf. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation this method sets the reaction constant in all tetrahedral elements of the compartment to kf
Note: The default value still comes from the steps.model description, so calling reset() will return the reaction constant to that value.
Syntax:
setCompReacK(comp, reac, kf)
Arguments: string comp string reac float kf
Return: None
Patch Data Access
- getPatchCount(p, s)
DEPRECATED Returns the number of molecules of species with identifier string spec in patch with identifier string patch.Note: in a mesh-based simulation this is the combined count from all triangular elements in the patch.
Syntax:
getPatchCount(patch, spec)
Arguments: string patch string spec
Return: float
- setPatchCount(p, s, n)
DEPRECATED Sets the number of molecules of species with identifier string spec in patch with identifier string patch to n. Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
setPatchCount(patch, spec, n)
Arguments: string patch string spec int n
Return: float
- getPatchArea(p)
Returns the area of patch with identifier string patch (in m^2).
Syntax:
getPatchArea(patch)
Arguments: string patch
Return: float
- getPatchAmount(p, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in patch with identifier string patch.
Note: in a mesh-based simulation this is the combined amount from all triangular elements in the patch.
Syntax:
getPatchAmount(patch, spec)
Arguments: string patch string spec
Return: float
- setPatchAmount(p, s, a)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in patch with identifier string patch to a. In a discrete solver, such as Wmdirect and Tetexact, this continuous value is converted internally into a discrete number of molecules by multiplication with Avogadro’s number.
Note: in a mesh-based simulation the molecules are divided as equally as possible over all triangular elements in the patch (i.e. a uniform distribution).
Syntax:
setPatchAmount(patch, spec, a)
Arguments: string patch string spec float a
Return: None
- setPatchSReacK(p, r, kf)
Sets the macroscopic reaction constant of surface reaction with identifier string sreac in patch with identifier string patch to kf. The unit of the reaction constant depends on the order of the reaction.
Note: In a mesh-based simulation this method sets the surface reaction constant in all triangular elements of the patch to kf.
Note: The default value still comes from the steps.model description, so calling reset() will return the surface reaction constant to that value.
Syntax:
setPatchSReacK(patch, reac, kf)
Arguments: string patch string reac float kf
Return: None
Tetrahedral Data Access
- getTetVol(idx)
Returns the volume (in m^3) of the tetrahedral element with index idx.
Syntax:
getTetVol(idx)
Arguments: index_t idx
Return: float
- getTetCount(idx, s)
DEPRECATED Returns the number of molecules of species with identifier string spec in the tetrahedral element with index idx.
Syntax:
getTetCount(idx, spec)
Arguments: index_t idx string spec
Return: int
- setTetCount(idx, s, n)
DEPRECATED Sets the number of molecules of species with identifier string spec in tetrahedral element with index idx to n.
Syntax:
setTetCount(idx, spec, n)
Arguments: index_t idx string spec int n
Return: None
- getTetAmount(idx, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in tetrahedral element with index idx.
Syntax:
getTetAmount(idx, spec)
Arguments: index_t idx string spec
Return: float
- setTetAmount(idx, s, m)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in tetrahedral element with index idx to a. This continuous value must be converted internally to a discrete number of molecules by multiplication with Avogadro’s number.
Due to the small volumes of tetrahedral elements the difference between ‘rounding up’ and ‘rounding down’ can be a significant difference in concentration.
Syntax:
setTetAmount(idx, spec, a)
Arguments: index_t idx string spec float a
Return: None
- getTetConc(idx, s)
DEPRECATED Returns the concentration (in Molar units) of species with identifier string spec in a tetrahedral element with index idx.
Syntax:
getTetConc(idx, spec)
Arguments: index_t idx string spec
Return: float
- setTetConc(idx, s, c)
DEPRECATED Sets the concentration (in Molar units) of species with identifier string spec in a tetrahedral element with index idx to conc.This continuous value must be converted internally to a discrete number of molecules.
Due to the small volumes of tetrahedral elements the difference between ‘rounding up’ and ‘rounding down’ can be a large difference in concentration.
Syntax:
setTetConc(idx, spec, conc)
Arguments: index_t idx string spec float conc
Return: None
- setTetReacK(idx, r, kf)
Sets the macroscopic reaction constant of reaction with identifier string reac in tetrahedral element with index idx to kf. The units of the reaction constant depends on the order of the reaction.
Syntax:
setTetReacK(idx, reac, kf)
Arguments: index_t idx string reac float kf
Return: None
- getTetV(idx)
Returns the potential (in volts) of tetrahedral element with index idx, taken at the barycenter.
Syntax:
getTetV(idx)
Arguments: index_t idx
Return: float
- setTetV(idx, v)
Set the potential (in volts) of tetrahedral element with index idx.
Syntax:
setTetV(idx, v)
Arguments: index_t idx float v
Return: None
- getTetVClamped(idx)
Returns true if the potential of tetrahedral element with index idx is clamped to some voltage.
Syntax:
getTetVClamped(idx)
Arguments: index_t idx
Return: bool
- setTetVClamped(idx, cl)
Sets whether the potential of tetrahedral element with index idx is clamped (clamped = True) or not (clamped = False).
Syntax:
setTetVClamped(idx, clamped)
Arguments: index_t idx bool clamped
Return: None
Triangular Data Access
- getTriArea(idx)
Returns the area (in m^2) of the triangular element with index idx.
Syntax:
getTriArea(idx)
Arguments: index_t idx
Return: float
- getTriCount(idx, s)
DEPRECATED Returns the number of molecules of species with identifier string spec in the triangular element with index idx.
Syntax:
getTriCount(idx, spec)
Arguments: index_t idx string spec
Return: float
- setTriCount(idx, s, n)
DEPRECATED Sets the number of molecules of species with identifier string spec in triangular element with index idx to n.
Syntax:
setTriCount(idx, spec, n)
Arguments: index_t idx string spec int n
Return: None
- getTriAmount(idx, s)
DEPRECATED Returns the amount (in mols) of species with identifier string spec in triangular element with index idx.
Syntax:
getTriAmount(idx, spec)
Arguments: index_t idx string spec
Return: float
- setTriAmount(idx, s, m)
DEPRECATED Sets the amount (in mols) of species with identifier string spec in triangular element with index idx to a. This continuous value must be converted internally to a discrete number of molecules by multiplication with Avogadro’s number.
Syntax:
setTriAmount(idx, spec, a)
Arguments: index_t idx string spec float a
Return: None
- setTriSReacK(idx, r, kf)
Sets the macroscopic reaction constant of surface reaction with identifier string sreac in triangular element with index idx to kf. The units of the reaction constant depends on the order of the reaction.
Syntax:
setTriSReacK(idx, reac, kf)
Arguments: index_t idx string reac float kf
Return: None
- getTriV(idx)
Returns the potential (in volts) of triangle element with index idx, taken at the barycenter.
Syntax:
getTriV(idx)
Arguments: index_t idx
Return: float
- setTriV(idx, v)
Set the potential (in volts) of triangle element with index idx.
Syntax:
setTriV(idx, v)
Arguments: index_t idx float v
Return: None
- getTriVClamped(idx)
Returns true if the potential of triangle element with index idx is clamped to some voltage.
Syntax:
getTriVClamped(idx)
Arguments: index_t idx
Return: bool
- setTriVClamped(idx, cl)
Sets whether the potential of triangle element with index idx is clamped (clamped = True) or not (clamped = False).
Syntax:
setTriVClamped(idx, clamped)
Arguments: index_t idx bool clamped
Return: None
- setTriIClamp(idx, i)
Set current clamp to triangle element with index idx to current i (amps). NOTE: Convention is maintained that a positive current clamp is depolarizing, a negative current clamp is hyperpolarizing.
Syntax:
setTriIClamp(idx, i)
Arguments: index_t idx float i
Return: None
- getTriI(idx)
Returns the current of triangle element with index idx, in amps, at the last EField calculation step.
Syntax:
getTriI(idx)
Arguments: index_t idx
Return: float
Vertex Data Access
- getVertV(vidx)
Returns the potential (in volts) of vertex element with index idx.
Syntax:
getVertV(idx)
Arguments: index_t idx
Return: float
- setVertV(vidx, v)
Set the potential (in volts) of vertex element with index idx.
Syntax:
setVertV(idx, v)
Arguments: index_t idx float v
Return: None
- getVertVClamped(vidx)
Returns true if the potential of vertex element with index idx is clamped to some voltage.
Syntax:
getVertVClamped(idx)
Arguments: index_t idx
Return: bool
- setVertVClamped(vidx, cl)
Sets whether the potential of vertex element with index idx is clamped (clamped = True) or not (clamped = False).
Syntax:
setVertVClamped(idx, clamped)
Arguments: index_t idx bool clamped
Return: None
- setVertIClamp(vidx, i)
Set current clamp to vertex element with index idx to current i (Amps). NOTE: Convention is maintained that a positive current clamp is depolarizing, a negative current clamp is hyperpolarizing.
Syntax:
setVertIClamp(idx, i)
Arguments: index_t idx float i
Return: None
Membrane Data Access
- setMembPotential(m, v)
Sets the potential (in volts) of membrane with string identifier memb. NOTE: This method will set the potential of all nodes in the volume conductor to the same value.
Syntax:
setMembPotential(memb, v)
Arguments: string memb float v
Return: None
- setMembCapac(m, cm)
Sets the specific membrane capacitance (in farad / m^2) of membrane with string identifier memb.
Syntax:
setMembCapac(memb, cm)
Arguments: string memb float cm
Return: None
- setMembVolRes(m, ro)
Sets the bulk electrical resistivity (in ohm.m) of the volume conductor assocaited with membrane with string identifier memb.
Syntax:
setMembVolRes(memb, ro)
Arguments: string memb float ro
Return: None
- setMembRes(m, ro, vrev)
Sets the surface electrical resistivity ro (in ohm.m^2) of the membrane with string identifier memb. Reversal potential vrev is required in Volts.
Syntax:
setMembRes(memb, ro, vrev)
Arguments: string memb float ro float vrev
Return: None
CVODE functions
- setTolerances(atol, rtol)
Set the absolute tolerance and the relative tolerance for CVODE.
Syntax:
setTolerance(atol, rtol)
Arguments: float atol float rtol
Return: None
- setMaxNumSteps(maxn)
Sets the maximum number of steps in CVODE per call to run(). Default is 10000 if this function is not called.
Syntax:
setMaxNumSteps()
Arguments: int maxn
Return: None